Adsorpcja zachodzi na granicy faz. Dlatego zasadne jest uznanie termodynamicznego opisu zjawisk powierzchniowych za szczególny przypadek termodynamiki układów heterogenicznych.
Ryż. 3.4. Adsorpcja Gibbsa: 1- dwufazowy układ porównawczy, 2- rzeczywisty układ dwufazowy z niejednorodnym obszarem
Znajduje zastosowanie w termodynamice układów heterogenicznych zasada addytywności co jest następujące: wszystkie rozległe właściwości układu heterogenicznego są równe sumie odpowiednich rozległych właściwości, jakie miałyby fazy przed ich zetknięciem. Oznaczmy fazy przez α i β (ryc. 4). Następnie dla układu idealnego, w którym właściwości faz w pobliżu granicy faz pokrywają się z ich właściwościami masowymi, obowiązują następujące zależności dla energii wewnętrznej U, objętości V, masy (liczby moli) n, entropii S po ustaleniu równowagi w system heterogeniczny:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Zakłada się, że temperatura i ciśnienie w obu fazach są takie same.
W przypadku rzeczywistych układów heterogenicznych obszar przejściowy na granicy dwóch faz wnosi dodatkowy wkład do rozległych właściwości układu. Jeżeli występują zjawiska powierzchniowe, należy wziąć pod uwagę różnicę między ekstensywnymi właściwościami rzeczywistego układu heterogenicznego a ekstensywnymi właściwościami układu modelowego, w którym nie występują zjawiska powierzchniowe. Taki system nazywa się systemem porównawczym. Układ porównawczy ma te same parametry intensywne (T, P, C i...) i taką samą objętość V jak układ rzeczywisty (rys. 4).
Z termodynamicznego punktu widzenia przez wielkość adsorpcji G rozumie się nadmiarową ilość substancji n s, wyrażoną w molach lub gramach, jaką posiada rzeczywisty układ heterogeniczny w porównaniu z układem odniesienia, w odniesieniu do powierzchni międzyfazowej lub pola powierzchni adsorbentu A. Zakłada się, że układ porównawczy ma te same parametry intensywne (T, P, C i) i tę samą objętość (V = V α + V β) co układ rzeczywisty (rys. 4) .
Г = (n - n α - n β)/A = n s /A 3.11
Nadmiarowe funkcje termodynamiczne obszaru przejściowego układu rzeczywistego (oznaczamy je indeksem s) można zapisać jako
U s = U - U α - U β , n s = n - n α - n β , S s \u003d S - S α - S β itp.
Eksperymentalne pomiary adsorpcji zawsze dają adsorpcję dokładnie jako nadmiar składnika w układzie rzeczywistym w porównaniu z wybranym układem odniesienia. Przykładowo przy adsorpcji gazu na adsorbencie stałym lub przy adsorpcji składników na fazie stałej, aby znaleźć wartości adsorpcji, wyznacz zmianę początkowych stężeń adsorbatu po zetknięciu faz α i β
n ja s = V(C ja o - C ja),
Gdzie C i o– początkowe stężenie i-tego składnika, C ja– stężenie i-tego składnika po ustaleniu równowagi pomiędzy stykającymi się fazami. Uważa się, że objętość V nie zmienia. Jednak koncentracja I komponent C ja, otrzymany doświadczalnie, określa się objętościowo V' powyżej granicy faz bez uwzględnienia objętości niejednorodnego obszaru warstwy przejściowej Vα na granicy faz, gdzie występuje stężenie C i α. Zatem ze względu na istnienie niejednorodnego obszaru w układzie rzeczywistym całkowitą objętość układu można przedstawić jako V = V’ + V α. Wszystkie ilości I-ty składnik C i o zostaną rozdzielone pomiędzy te dwa tomy:
V do ja o = V’ do ja + V α do ja α ,
i liczbę moli składnika I, zaadsorbowany na granicy faz, będzie równy
n ja s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Te. wyznaczona eksperymentalnie adsorpcja to nadmiar i-tego składnika w objętości V α w porównaniu z ilością tego składnika w tej samej objętości, daleko od granicy faz. Ten typ adsorpcji nazywa się adsorpcją Gibbsa. .
V α C i α nazywana pełną treścią I- składnik warstwy adsorpcyjnej. W obszarze bardzo niskich stężeń C ja w objętości V' poprawka V α C ja równanie (3.2) można pominąć i uwzględnić zmierzoną wartość V α C i α pełna treść I- składnika warstwy adsorpcyjnej, na przykład podczas adsorpcji gazu na stałym adsorbencie pod niskim ciśnieniem.
Termodynamika procesów adsorpcji.
| Nazwa parametru | Oznaczający |
| Temat artykułu: | Termodynamika procesów adsorpcji. |
| Rubryka (kategoria tematyczna) | Edukacja |
Podstawowe definicje i metody klasyfikacji procesów adsorpcji.
Adsorpcja odnosi się do zjawisk zachodzących w wyniku spontanicznego spadku energii powierzchniowej.
Adsorpcja– proces samoistnej odwracalnej lub nieodwracalnej redystrybucji składników układu heterogenicznego pomiędzy warstwą powierzchniową a objętością fazy jednorodnej.
W układach wieloskładnikowych korzystnie na warstwę powierzchniową przenosi się składnik, który silniej zmniejsza napięcie międzyfazowe. W układach jednoskładnikowych podczas tworzenia warstwy powierzchniowej następuje zmiana jej struktury (pewna orientacja atomów i cząsteczek, polaryzacja), zwana autoadsorpcja.
Faza gęstsza, na której zlokalizowane są oddziaływania adsorpcyjne, nazywana jest fazą gęstszą adsorbent. Substancję rozdzieloną pomiędzy objętość fazy jednorodnej a warstwę powierzchniową oznaczono terminem „” adsorbatʼʼ.
W niektórych przypadkach proces adsorpcji jest odwracalny. W takim przypadku pod pewnymi warunkami część zaadsorbowanych cząsteczek w wyniku molekularnych zjawisk kinetycznych może przejść z warstwy powierzchniowej do fazy objętościowej. Odwrotny proces adsorpcji nazywa się desorpcja.
Metody klasyfikacji procesów adsorpcji.
Klasyfikacja procesów adsorpcji ze względu na stan skupienia oddziałujących faz. Biorąc pod uwagę zależność od stanu skupienia faz sąsiednich, wyróżnia się następujące typy procesów adsorpcji:
Adsorpcja gazów na stałych adsorbentach;
Adsorpcja substancji rozpuszczonych na granicy faz „ciało stałe-ciecz” i „ciecz-ciecz”;
Adsorpcja środków powierzchniowo czynnych na granicy faz ciecz-gaz.
Klasyfikacja procesów adsorpcji ze względu na mechanizm oddziaływania adsorbentu i adsorbatu. Adsorpcję można rozpatrywać jako oddziaływanie cząsteczek adsorbatu z centrami aktywnymi adsorbentu. Zgodnie z mechanizmem ich interakcji dzieli się następujące rodzaje adsorpcji:
1) adsorpcja fizyczna (molekularna).– oddziaływanie cząsteczek adsorbatu z adsorbentem odbywa się pod wpływem sił van der Waalsa, wiązań wodorowych (bez reakcji chemicznych);
2) adsorpcja chemiczna (chemisorpcja)– przyłączenie cząsteczek adsorbatu do centrów aktywnych adsorbentu następuje w wyniku różnego rodzaju reakcji chemicznych (z wyjątkiem reakcji wymiany jonowej);
3) Adsorpcja jonowymienna (wymiana jonowa) – redystrybucja substancji adsorbowanej pomiędzy roztworem a fazą stałą (wymiennik jonowy) zgodnie z mechanizmem reakcji wymiany jonowej.
Aby ilościowo opisać procesy adsorpcji, stosuje się dwie wielkości.
1) Absolutna adsorpcja– ilość (mol) lub masa (kg) adsorbatu na jednostkę powierzchni lub masę adsorbentu. Oznaczenie – A; wymiar: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Adsorpcja Gibbsa (nadmiarowa).– nadmiar substancji adsorbatu w warstwie powierzchniowej o określonej grubości w stosunku do jej ilości w objętości fazy jednorodnej na jednostkę powierzchni lub masy adsorbentu. Oznaczenie – G; wymiar: mol/m 2, mol/kᴦ.
Zależność pomiędzy adsorpcją bezwzględną i nadmiarową można zilustrować równaniem:
Г = А – с * h (3.1)
gdzie c jest równowagowym stężeniem substancji w objętości fazy, mol/m3;
h jest grubością warstwy wierzchniej, umownie przyjmowaną jako 10 -9 m.
W wieloskładnikowych układach heterogenicznych, gdy jeden lub drugi składnik jest redystrybuowany pomiędzy objętością fazy jednorodnej a warstwą powierzchniową, obowiązuje równanie na nadmiar energii wewnętrznej powierzchni:
U = T * S + s * s + Sm i * n ja (3.2)
Redukując wszystkie wyrazy równania do jednostkowego pola powierzchni międzyfazowej, otrzymujemy:
U s = T * S s + s + Sm i * Г ja (3.3)
gdzie Г i = n i / s jest nadmiarem i-tego składnika w warstwie powierzchniowej, czyli adsorpcją Gibbsa.
Dla układu jednoskładnikowego równanie (3.3) będzie miało postać:
G s = s + m * G (3.4)
gdzie G s = U s - T * S s – energia Gibbsa powierzchni lub praca tworzenia jednostkowej powierzchni;
m * G – zagęszczenie substancji zaadsorbowanej w warstwie powierzchniowej.
Na podstawie równania (3.4) można stwierdzić, że podczas adsorpcji praca tworzenia powierzchni międzyfazowej polega na pracy tworzenia powierzchni (rozerwaniu wiązań kohezyjnych w objętości fazy adsorbatu) i zagęszczeniu substancji w warstwie powierzchniowej.
W stanie równowagi dynamicznej pomiędzy adsorbentem a adsorbatem, zmiana energii Gibbsa układu heterogenicznego ΔG = 0, termodynamikę procesu adsorpcji opisuje równanie zwane Podstawowe równanie adsorpcji Gibbsa:
Ds = S° i * dm i (3,5)
Równanie to jest uniwersalne, gdyż obowiązuje dla wszystkich typów procesów adsorpcji
Szczególne przypadki równania adsorpcji Gibbsa.
1) Adsorpcja z roztworów.
Dla potencjału chemicznego i-tego składnika układu podczas adsorpcji na granicy faz „ciecz – ciało stałe” i „ciecz – gaz” obowiązują następujące równania:
m ja = m ja 0 + R*T*ln a ja (3.6)
dm i = R*T* d ln a i (3.7)
gdzie m i 0 jest potencjałem chemicznym i-tego składnika układu w warunkach standardowych;
a i jest aktywnością i-tego składnika systemu w warunkach standardowych.
Na tej podstawie równanie adsorpcji Gibbsa przyjmuje postać:
Г i = - a i / R*T * (ds / da i) (3.8)
Dla roztworów nieelektrolitów bierzemy a i = c i, wówczas:
Г i = - с / R*T * (ds / dс) (3.9)
Dla roztworów elektrolitów:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
gdzie с ± jest średnim stężeniem jonowym roztworu;
n jest współczynnikiem stechiometrycznym.
2) Adsorpcja substancji z fazy gazowej.
Zgodnie z równaniem Mendelejewa-Clayperona:
Р = с * R*T (3.11)
W związku z tym równanie Gibbsa dotyczące adsorpcji gazów na stałych adsorbentach zapisuje się w następującej formie:
Г i = - Р / R*T * (ds / dР) (3.12)
W praktyce równanie adsorpcji Gibbsa pozwala na podstawie pomiarów napięcia powierzchniowego przy różnych wartościach stężenia cieczy lub równowagowego ciśnienia gazu obliczyć wielkość adsorpcji substancji w warstwie międzyfazowej, dla której wyznacza się napięcie powierzchniowe.
Termodynamika procesów adsorpcji. - koncepcja i rodzaje. Klasyfikacja i cechy kategorii „Termodynamika procesów adsorpcji”. 2017, 2018.
Adsorpcji jako spontanicznemu stężeniu cząsteczek na powierzchni towarzyszy spadek entropii układu. Ponieważ kryterium spontaniczności procesu jest
∆H - T · ∆S = ∆G< 0,
wówczas adsorpcja jest możliwa tylko przy ∆H< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Wraz ze wzrostem temperatury równowaga przesuwa się w stronę procesu endotermicznego, czyli desorpcji.
Adsorpcja na stałej powierzchni
1. Adsorpcja monocząsteczkowa.
Zgodnie z teorią Langmuira cząsteczki adsorbentu oddziałują z powierzchnią adsorbentu, tworząc ostatecznie warstwę monocząsteczkową. W tym przypadku stopień wypełnienia () powierzchni zaadsorbowaną substancją podczas adsorpcji z fazy gazowej
z płynu
gdzie K jest stałą równowagi (stała adsorpcji);
p jest ciśnieniem cząstkowym zaadsorbowanego gazu;
c jest stężeniem zaadsorbowanej substancji.
Zależność β od p (lub c) przedstawia wykres (izoterma adsorpcji, T = const) na ryc. 1.3.

Ryż. 1.3. Stopień wypełnienia powierzchni zaadsorbowaną substancją
Przy niskich stężeniach i ciśnieniach cząstkowych adsorpcja jest proporcjonalna do stężenia lub ciśnienia cząstkowego:
R<< 1, β ≈ К· r ilis<< 1, β ≈ К· s, tj. początkowy odcinek izotermy jest w przybliżeniu liniowy i tan α = K (tg α jest określane przez nachylenie krzywej w punkcie p (lub c) → 0: lub ).
If to liczba moli zaadsorbowanej substancji na 1 g adsorbentu; - maksymalną możliwą liczbę moli zaadsorbowanej substancji na 1 g adsorbentu („pojemność jednowarstwy”), wówczas
Podstawiając β do równania (1.3) (w przypadku adsorpcji z fazy gazowej stężenie Z w równaniach należy zastąpić ciśnieniem R), otrzymujemy:
 (1.6)
(1.6)
Ponieważ i K w danej parze adsorbent-adsorbent są stałymi (at T=const), to według zależności można znaleźć DO(ryc. 1.4).

Ryż. 1.4. Graficzne rozwiązanie równania adsorpcji
otrzymane przez ekstrapolację eksperymentalnej zależności liniowej na () = 0; i od tego czasu , .
Wartość można wykorzystać do określenia powierzchni właściwej adsorbentu UD (w m 2 na 1 g adsorbentu), jeżeli znana jest powierzchnia ω zajmowana na powierzchni przez jedną cząsteczkę adsorbentu (wyznaczona na podstawie wielkości cząsteczki):
UD = · ω · Nie, (1,7)
gdzie Na jest liczbą Avogadra (Na = 6,02 · 10 · 23).
Z kolei znaną wartość UD można obliczyć ω dowolnej substancji na podstawie jej adsorpcji na danym adsorbencie.
2. Adsorpcja wielocząsteczkowa.
Równanie (1.5) opisuje krzywą z nasyceniem, tj. Na
p (lub c) → ∞ dąży do wartości granicznej równej (ryc. 1.5,a).

Ryc.1.5. Izotermy adsorpcji:
a – adsorpcja z nasyceniem; b – adsorpcja wielocząsteczkowa
Jednak w niektórych przypadkach izotermy adsorpcji wyglądają jak te pokazane na ryc. 1,5, b, tj. nie osiąga granicy nawet przy wysokim p (lub c).
Zależności typu pokazane na ryc. 1.5,b odpowiadają adsorpcji wielocząsteczkowej. Z reguły takie izotermy są charakterystyczne dla substancji o silnych oddziaływaniach międzycząsteczkowych (na przykład wody). Kiedy centra adsorpcyjne na powierzchni adsorbentu zostaną zajęte (warstwa jednocząsteczkowa zostanie nasycona), następuje „lądowanie” kolejnych cząsteczek adsorbatu w wyniku oddziaływań międzycząsteczkowych z cząsteczkami już zaadsorbowanymi (rys. 1.6). Ciepło takiej adsorpcji ma zbliżoną wartość bezwzględną, ale znak przeciwny do ciepła parowania odpowiedniej cieczy (zastanów się dlaczego).

Ryc.1.6. Schemat adsorpcji:
a - adsorpcja monocząsteczkowa; b - adsorpcja wielocząsteczkowa
Gdy się zbliżymy R do ciśnienia pary nasyconej zaadsorbowanej substancji zaczyna ona skraplać się na powierzchni adsorbentu, w efekcie szybko rośnie wraz ze wzrostem R.
W przypadku oddziaływania dwóch atomów:
U – energia oddziaływania;
U = U PRZED. + POWRÓT
 - Równanie Lennarda-Jonesa
, do, b, m = stała
- Równanie Lennarda-Jonesa
, do, b, m = stała
W przypadku oddziaływania atomów z powierzchnią stałą konieczne jest zsumowanie wszystkich oddziaływań.
x – odległość od powierzchni
r – promień działania sił przyciągających
dV – objętość
n – liczba cząsteczek powierzchniowych
U REKLAMY. – energia oddziaływania adsorpcyjnego



W przypadku adsorpcji przyciąganie wzrasta. Natomiast w przypadku oddziaływania niepolarno-niepolarnego, adsorpcja jest zlokalizowana głównie w zagłębieniach.
Oddziaływanie elektrostatyczne.
Adsorbent polarny – adsorbat niepolarny
Adsorbent niepolarny – adsorbat polarny
Adsorbent polarny – adsorbat polarny.
M  Cząsteczka adsorbatu jest reprezentowana jako dipol, a adsorbent jako przewodnik, w którym cząsteczka adsorbatu indukuje zwierciadło dipolowe symetrycznie względem danego.
Cząsteczka adsorbatu jest reprezentowana jako dipol, a adsorbent jako przewodnik, w którym cząsteczka adsorbatu indukuje zwierciadło dipolowe symetrycznie względem danego.
X – odległość do środka
Podczas interakcji pojawia się potencjał:
 ,
,
 - moment dipolowy.
- moment dipolowy.
Potencjał ma tendencję do przyjmowania wartości maksymalnej, tj. dipole mają tendencję do ustawiania się prostopadle do powierzchni.
Ponieważ wzrost temperatury sprzyja wzrostowi ruchów Browna, prowadzi to do zahamowania procesu adsorpcji.
W przypadku oddziaływania elektrostatycznego adsorbat lokalizuje się głównie na występach.
Podstawowe równanie adsorpcji.
W przypadku adsorpcji następuje redystrybucja składnika, co oznacza zmianę potencjału chemicznego. Proces adsorpcji można uznać za przemianę energii powierzchniowej w energię chemiczną.
Objętość warstwy = 0, następnie uogólnione równanie I i II zasady termodynamiki:
T = stała; (1) = (2) => 

Dla układu dwuskładnikowego:
,
 ,
,
 =>
=>
=>
 - Równanie adsorpcji Gibbsa
.
- Równanie adsorpcji Gibbsa
.
W przypadku adsorpcji telewizyjnej. korpus - gaz: , 
 ,
,

 - izoterma
- izoterma
 - izobar
- izobar
 - izopiknalny
- izopiknalny
 - izoster
- izoster
Izoterma, izopikne, izostera są ze sobą powiązane.
Ponieważ funkcja adsorpcji 



Izoterma Henry'ego Izoterma Langmuira



Termodynamika. Adsorpcja.
Dla substancji skondensowanej:
 ,
,
 ,
,
 - całkowa zmiana energii Gibbsa
.
- całkowa zmiana energii Gibbsa
.

P – nacisk na zakrzywioną powierzchnię, Р S – nacisk na płaską powierzchnię
 - potencjał adsorpcji
- potencjał adsorpcji
Różnicowa zmiana entrapii 
 , Г = stała
, Г = stała
- różnicowa zmiana entropii
- różnicowa entalpia adsorpcji
 - izosteryczne ciepło adsorpcji
- izosteryczne ciepło adsorpcji
 - ciepło kondensacji
- ciepło kondensacji
 - ciepło netto adsorpcji
- ciepło netto adsorpcji

 ,
,




Qa – całkowe ciepło adsorpcji, 
Qra – całkowe ciepło netto adsorpcji,

Równanie Henry'ego
Badanie adsorpcji komplikuje niejednorodność powierzchni, dlatego najprostsze prawa uzyskuje się dla powierzchni jednorodnych.
Rozważmy oddziaływanie gazów z powierzchnią stałą, gdy gaz przechodzi ze stanu równowagi objętościowej do stanu równowagi na powierzchni. Przypadek ten jest analogiczny do równowagi gazów w polu grawitacyjnym.
 ,
,
 ,
=>
,
=> -Równanie Henry'ego
-Równanie Henry'ego
 - współczynnik podziału
- współczynnik podziału
W procesie adsorpcji następuje zmiana potencjałów chemicznych.
Dla fazy masowej: 
Dla gazu na powierzchni: 
W stanie równowagi  , tj.
, tj.

W równaniu Henry'ego stała nie zależy od stężenia
Równanie Henry'ego obowiązuje w obszarze niskich ciśnień i stężeń. Wraz ze wzrostem stężenia możliwe są dwa rodzaje odstępstw od prawa Henry’ego:
1 – odchylenia dodatnie, D maleje, A maleje
2 – odchylenia ujemne, D – wzrosty, A – wzrosty.
Rodzaj odchylenia zależy od przewagi jednego lub drugiego rodzaju interakcji adsorbent-adsorbat.
Przy silnym oddziaływaniu kleju współczynniki aktywności rosną - odchylenie dodatnie. W przypadku oddziaływań kohezyjnych obserwuje się odchylenia ujemne.
Adsorpcja monocząsteczkowa.
Izoterma Langmuira.
Najprostsze wzory uzyskano w teorii Henry'ego. Langmuir zaproponował teorię, zgodnie z którą adsorpcję uważa się za reakcję quasi-chemiczną. W której:
Powierzchnia jest energetycznie jednorodna.
Adsorpcja jest zlokalizowana, każde centrum adsorpcji oddziałuje z jedną cząsteczką adsorbatu.
Cząsteczki adsorbatu nie oddziałują ze sobą.
Adsorpcja jednowarstwowa.

 - powierzchnia,
- powierzchnia,  - adsorbat,
- adsorbat,  - kompleks adsorpcyjny.
- kompleks adsorpcyjny.
 , to stężenie miejsc adsorpcji:
, to stężenie miejsc adsorpcji:  ,
, - ograniczenie adsorpcji.
- ograniczenie adsorpcji.
 , wówczas stała reakcji wynosi:
, wówczas stała reakcji wynosi: 

 - Równanie Langmuira.
- Równanie Langmuira.
Zależność adsorpcji od stężenia
1 )
)
 ,
,
2) obszar wysokich stężeń
 - ograniczenie adsorpcji, tworzenie warstwy monocząsteczkowej
- ograniczenie adsorpcji, tworzenie warstwy monocząsteczkowej
Dla energii Gibbsa: .
g jest współczynnikiem entropii.
W przypadku izotermy Henry'ego energia Gibbsa charakteryzuje przejście adsorbatu ze stanu standardowego w masie do stanu standardowego na powierzchni. W przypadku izotermy Langmuira  charakteryzuje stopień powinowactwa pomiędzy adsorbentem a adsorbatem.
charakteryzuje stopień powinowactwa pomiędzy adsorbentem a adsorbatem.
 znalezione z izobary van't Hoffa.
znalezione z izobary van't Hoffa.

 , Następnie
, Następnie  , stąd
, stąd  .
.

 - stopień wypełnienia powierzchni.
- stopień wypełnienia powierzchni.




 - ilość wolnych miejsc,
- ilość wolnych miejsc,  - liczba zajętych miejsc.
- liczba zajętych miejsc.
 ,
,

Te. w obszarze wysokich stężeń liczba wolnych miejsc jest odwrotnie proporcjonalna do ilości adsorbatu.
Adsorpcja mieszaniny gazów na jednorodnej powierzchni.
W tym przypadku proces adsorpcji uważa się za dwie równoległe reakcje.
 (1)
(1)

 (2)
(2)


Adsorpcja mieszaniny gazów na niejednorodnej powierzchni.
W przypadku niejednorodnej powierzchni nie można ograniczać się do wypełnień przeciętnych.
W wyniku konkurencji możliwa jest lokalizacja różnych adsorbatów na obszarach różnego typu.
W tym wypadku relacja  .
.
 ,
,
 - prężność pary nasyconej adsorbatu.
- prężność pary nasyconej adsorbatu.
 ,
,
 - ciepło adsorpcji.
- ciepło adsorpcji.
„+” – zależność od symbatu, „-” – zależność od antybatu, „N” – brak korelacji.
„+” - adsorpcja przebiega według tego samego mechanizmu. W obszarach najbardziej korzystnych energetycznie adsorbowany jest głównie gaz o dużym powinowactwie do powierzchni.
„-” - adsorpcja zachodzi poprzez różne mechanizmy i do pewnego momentu nie ma już konkurencji o powierzchnię.
Adsorpcja monocząsteczkowa realizowana jest głównie podczas fizycznej adsorpcji gazów przy niskich wartościach P, jak również na granicy faz ciecz/gaz.
Adsorpcja wielocząsteczkowa.
Teoria zakładu(Brunauer, Emmett, Teller).
W przypadku, gdy utworzenie monowarstwy nie jest wystarczające do skompensowania energii powierzchniowej, adsorpcja jest wielocząsteczkowa i można ją rozpatrywać w wyniku wymuszonej kondensacji pod działaniem sił powierzchniowych.
Kluczowe punkty:
Kiedy cząsteczka adsorbatu uderza w zajęte miejsce, powstaje zestaw wielokrotny.
Gdy się zbliżymy P Do P S zmniejsza się liczba miejsc wolnej adsorpcji. Początkowo liczba miejsc zajmowanych przez osoby samotne, podwójne itp. wzrasta, a następnie maleje. w zestawach.
Na P =P S adsorpcja zamienia się w kondensację.
Nie ma interakcji poziomych.
Dla pierwszej warstwy spełniona jest izoterma Langmuira.
Powierzchnię traktuje się jako zbiór miejsc adsorpcji. Obowiązuje warunek równowagi dynamicznej: szybkość kondensacji w wolnych miejscach jest równa szybkości parowania z miejsc zajętych.
a jest współczynnikiem kondensacji (ułamek cząsteczek skondensowanych na powierzchni);
 ,
,
Zm – maksymalna liczba wolnych miejsc.
 - częstotliwość drgań atomu w kierunku prostopadłym do powierzchni.
- częstotliwość drgań atomu w kierunku prostopadłym do powierzchni.
Dla pierwszej warstwy warunki równowagi dynamicznej:



 , Następnie
, Następnie
 - Równanie Langmuira.
- Równanie Langmuira.
Dla drugiej warstwy będzie to prawdą: 
Dla i-tej warstwy: 
Dla uproszczenia zakłada się, że a i ν są takie same dla wszystkich warstw z wyjątkiem pierwszej. Dla wszystkich warstw z wyjątkiem pierwszej ciepło adsorpcji jest stałe. Dla ostatniej warstwy ciepło adsorpcji jest równe ciepłu kondensacji. W rezultacie otrzymano równanie
 (*)
(*)
C– stała,
W przypadku teorii BET stała Z charakteryzuje energię Gibbsa czystej adsorpcji. Równanie zawiera tylko jedną stałą i równanie to jest również bardzo ważne przy określaniu powierzchni właściwej adsorbentu.
Ponieważ w wyniku adsorpcji wydziela się ciepło, w niskich temperaturach określa się powierzchnię właściwą.
 ????????????
????????????


Główna wada teorii– zaniedbanie oddziaływań poziomych na rzecz oddziaływań pionowych.
Równanie mieści się w przedziale  od 0,05 do 0,3.
od 0,05 do 0,3.
Gdzie  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – ma to wpływ na interakcję adsorbat – adsorbat.
> 0,3 – ma to wpływ na interakcję adsorbat – adsorbat.
Uwzględnianie oddziaływań adsorbat-adsorbat.
Interakcje zachodzą, gdy rozgałęzione cząsteczki lub cząsteczki są adsorbowane na niepolarnej powierzchni. Potrafi tworzyć współpracowników. W tym przypadku zmienia się kształt izoterm adsorpcji.
A  adsorbent nie jest polarny.
adsorbent nie jest polarny.
Wykres 1 przedstawia słabe oddziaływania adsorbat-adsorbat i silne oddziaływania adsorbat-adsorbent.
Wykres 2 przedstawia silne interakcje adsorbat-adsorbat i silne oddziaływania adsorbat-adsorbent.
Wykres 3 przedstawia silne oddziaływanie adsorbat-adsorbat i słabe oddziaływanie adsorbat-adsorbent.
 ,
,

W przypadku interakcji pomiędzy cząsteczkami adsorbatu należy uwzględnić zmiany współczynników aktywności. A to równanie zapisuje się jako:
 - Równanie Frunkina, Fowlera, Guggenheima.
- Równanie Frunkina, Fowlera, Guggenheima.
k– stała przyciągania.
Teoria potencjału Polyany’ego.
Teoria ta nie wyprowadza żadnego rodzaju izotermy adsorpcji, ale umożliwia obliczenie izoterm w innej temperaturze.
Adsorpcja- jest to wynik przyciągania adsorbatu do powierzchni adsorbentu na skutek działania potencjału adsorpcyjnego, który nie zależy od obecności innych cząsteczek, a zależy od odległości powierzchni od cząsteczki adsorbatu.
 ,
,
 - potencjał adsorpcji.
- potencjał adsorpcji.
Ponieważ powierzchnia nie jest jednorodna, odległość zastępuje się objętością adsorpcji  .Objętość adsorpcji jest objętością zawartą pomiędzy powierzchnią a punktem odpowiadającym danej wartości
.Objętość adsorpcji jest objętością zawartą pomiędzy powierzchnią a punktem odpowiadającym danej wartości  .
.
Potencjał adsorpcji to praca przeniesienia 1 mola adsorbatu poza zadaną objętość adsorpcji do zadanego punktu objętości adsorpcji (lub praca przeniesienia 1 mola pary nasyconej adsorbatu będącego w równowadze z ciekłym adsorbatem przy braku adsorbentu w fazę gazową pozostającą w równowadze z adsorbentem).

Krzywa charakterystyczna
 - potencjał adsorpcji,
- potencjał adsorpcji, 
Dla danego adsorbentu i różnych adsorbatów prawdziwe jest stwierdzenie: 
Dla różnych typów adsorbatów  ,
,
Gdzie  potencjały izoterm adsorpcji przy ciśnieniach względnych
potencjały izoterm adsorpcji przy ciśnieniach względnych  dla adsorbatu 1 i dla adsorbatu 2. Stosunek ten jest wartością stałą.
dla adsorbatu 1 i dla adsorbatu 2. Stosunek ten jest wartością stałą.
 - współczynnik powinowactwa
- współczynnik powinowactwa
Teoria kondensacji kapilarnej.
Przebieg procesu adsorpcji w dużej mierze zależy od budowy ciała porowatego.
|
Mikroporowaty | |
|
Przejściowy porowaty | |
|
Makroporowaty |
W przypadku sorbentów mikroporowatych pola sił adsorpcji nakładają się. W przypadku sorbentów makroporowatych pory pełnią rolę kanałów transportowych. Procesy kondensacji są najbardziej znaczące w ciałach przejściowo porowatych. Kondensacja kapilarna rozpoczyna się przy pewnych wartościach P I  , gdy część energii powierzchniowej została już skompensowana. Warunkiem koniecznym jest to, aby powierzchnia była samozwilżająca. Proces jest opisany Równanie Thompsona-Kelvina.
, gdy część energii powierzchniowej została już skompensowana. Warunkiem koniecznym jest to, aby powierzchnia była samozwilżająca. Proces jest opisany Równanie Thompsona-Kelvina.

 - w przypadku zwilżania środek krzywizny znajduje się w fazie gazowej.
- w przypadku zwilżania środek krzywizny znajduje się w fazie gazowej.
W przypadku kondensacji kapilarnej izoterma adsorpcji ma postać histeretyczną. Dolna gałąź odpowiada procesowi adsorpcji, a górna gałąź odpowiada procesowi desorpcji.
Wszystkie rodzaje porów można zredukować do trzech typów:
|
Stożkowy |
Cylindryczny z jednym zamkniętym końcem |
Cylindryczny z dwoma otwartymi końcami |
|
Wypełnianie procesowe odbywa się od dołu porów. Izoterma adsorpcji i izoterma desorpcji w tym przypadku pokrywają się, ponieważ proces adsorpcji rozpoczyna się od kuli, a proces desorpcji rozpoczyna się również wraz ze zniknięciem niektórych kul.
↓ |
Nie ma histerezy. Skok do przodu i do tyłu opisuje równanie:
|
Nigdzie nie ma dna, wypełnienie porów będzie przebiegać wzdłuż ścian cylindra.
cylinder: Izoterma będzie miała wygląd histeretyczny.
↓ |


 - kula,
- kula, ,
,



W  W warunkach zwilżania następuje kondensacja przy niższych ciśnieniach, co jest korzystne energetycznie. Z gałęzi desorpcji otrzymuje się krzywe rozkładu wielkości porów.
W warunkach zwilżania następuje kondensacja przy niższych ciśnieniach, co jest korzystne energetycznie. Z gałęzi desorpcji otrzymuje się krzywe rozkładu wielkości porów.
Maksimum krzywej różniczkowej jest przesunięte w lewo względem punktu przegięcia krzywej całkowej. Całkowita objętość małych porów jest niewielka, ale mają duże powierzchnie. Wraz ze wzrostem wielkości porów zwiększa się ich objętość  , a obszar jest podobny
, a obszar jest podobny  , z tego powodu obserwuje się przesunięcie maksimum krzywej różniczkowej.
, z tego powodu obserwuje się przesunięcie maksimum krzywej różniczkowej.
Adsorpcja na granicy faz ciało stałe-ciecz.
W przypadku adsorpcji na granicy faz ciało stałe-gaz zaniedbaliśmy jeden składnik. W przypadku adsorpcji na granicy faz ciało stałe-ciecz adsorbat wypiera cząsteczki rozpuszczalnika z powierzchni adsorbentu.
 ,
,
Równanie jest poprawne:

 ,
,
N 1, N 2 – ułamki molowe rozpuszczalnika i składnika, N 1 + N 2 = 1, wówczas
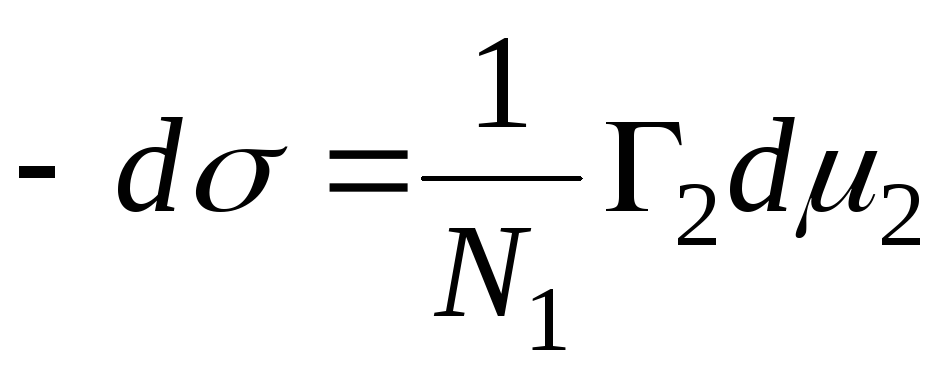 ,
=>
,
=>
 , to równanie adsorpcji dla granicy faz ciało stałe-ciecz.
, to równanie adsorpcji dla granicy faz ciało stałe-ciecz.
Adsorpcja (G) > 0 at  <
0
<
0
Jeśli wartości  dla składnika i rozpuszczalnika są bardzo różne, w tym przypadku zależność G z N ma ekstremum przy tej wartości N
~ 0,5.
dla składnika i rozpuszczalnika są bardzo różne, w tym przypadku zależność G z N ma ekstremum przy tej wartości N
~ 0,5.
mi  Jeśli
Jeśli  mają zbliżone wartości, w tym przypadku znak adsorpcji może się zmienić. Uzależnienie G z N przecina oś x
mają zbliżone wartości, w tym przypadku znak adsorpcji może się zmienić. Uzależnienie G z N przecina oś x
Punkt przecięcia funkcji G(N) z osią x nazywa się azeotrop adsorpcyjny. Oznacza to, że na danym adsorbencie nie da się rozdzielić tych dwóch składników.
Równanie izotermy adsorpcji ze stałą wymiany.
Podczas adsorpcji na granicy faz ciało stałe-ciecz następuje ciągła redystrybucja składników pomiędzy powierzchnią adsorbentu a objętością roztworu.
 - komponenty (- - odnoszą się do powierzchni)
- komponenty (- - odnoszą się do powierzchni)

 ,
,
 ,
, .
.
 ,
,


Adsorpcja na granicy faz ciecz-gaz
R  Rozważmy zmianę profilu stężenia w wyniku przecięcia granicy faz ciecz-gaz. Niech składnik 2 będzie lotny.
Rozważmy zmianę profilu stężenia w wyniku przecięcia granicy faz ciecz-gaz. Niech składnik 2 będzie lotny.
Cs – stężenie w warstwie powierzchniowej.
Na podstawie definicji nadmiernej adsorpcji

Jeżeli składnik nie jest lotny, wartość adsorpcji zostanie zapisana w następujący sposób:
P  ri
ri 

w równaniu  charakter substancji opisuje jej pochodna
charakter substancji opisuje jej pochodna  .
.
Izoterma napięcia powierzchniowego może mieć postać 1 lub 2:
1 – środki powierzchniowo czynne
2 – środki powierzchniowo czynne
Aktywność powierzchniowa g to zdolność substancji do zmniejszania napięcia powierzchniowego w układzie.

 - grubość warstwy wierzchniej
- grubość warstwy wierzchniej
C S– stężenie składnika w warstwie powierzchniowej
Z– stężenie objętościowe
Dla szeregu homologicznego obowiązuje zasada:
 - Rządy Traubo Duclosa
- Rządy Traubo Duclosa
Dla szeregu homologicznego izoterma adsorpcji wygląda następująco:
Zamiast A piszemy G, ponieważ adsorpcja w warstwie powierzchniowej jest nadmierna.
Izoterma napięcia powierzchniowego:

 - napięcie powierzchniowe czystego rozpuszczalnika.
- napięcie powierzchniowe czystego rozpuszczalnika.
 - podstawowe równanie adsorpcji;
- podstawowe równanie adsorpcji;
 - Równanie Langmuira.
- Równanie Langmuira.
Rozwiążmy je razem:

- Równanie Szyszkowskiego.
B– stała dla szeregu homologicznego.
A- przy przejściu z jednego homologu na drugi wzrasta 3-3,5 razy 
![]()
1 – obszar niskich stężeń
![]()
2 – stężenie średnie
3 – warstwa monocząsteczkowa
Surfaktanty to cząsteczki difilowe, tj. obejmują grupę polarną i niepolarny rodnik węglowodorowy.
o jest polarną częścią cząsteczki.
| - niepolarna część cząsteczki.
W rozpuszczalniku polarnym cząsteczki środka powierzchniowo czynnego są zorientowane w taki sposób, że polarna część cząsteczki jest zwrócona w stronę rozpuszczalnika, a część niepolarna jest wypychana do fazy gazowej.
W równaniu Szyszkowskiego  , jest stała dla szeregu homologicznego.
, jest stała dla szeregu homologicznego.
Zaczyna się pojawiać efekt środka powierzchniowo czynnego N>5. Przy stężeniach wyższych niż stężenie warstwy monocząsteczkowej w roztworach środków powierzchniowo czynnych następuje micelizacja.
Micele– nazywany jest agregatem cząsteczek amfifilowego środka powierzchniowo czynnego, którego rodniki węglowodorowe tworzą rdzeń, a grupy polarne przekształcają się w fazę wodną.
Masa micelarna – masa micelarna.
H  liczba cząsteczek – liczba agregacji.
liczba cząsteczek – liczba agregacji.
Micele sferyczne
W przypadku micelizacji w roztworze ustala się równowaga
CMC – stężenie krytyczne tworzenia miceli.
Ponieważ uważamy micelę za oddzielną fazę:
Dla szeregu homologicznego istnieje równanie empiryczne:
A– energia rozpuszczania grupy funkcyjnej.
B – przyrost potencjału adsorpcyjnego, praca adsorpcyjna na jednostkę metylenu.
– przyrost potencjału adsorpcyjnego, praca adsorpcyjna na jednostkę metylenu.
Obecność rdzenia węglowodorowego w micelach stwarza możliwość rozpuszczenia w wodnych roztworach środków powierzchniowo czynnych związków nierozpuszczalnych w wodzie, zjawisko to nazywa się solubilizacją (rozpuszcza się solubilizat, surfaktant jest solubilizatorem).
Błoto może być całkowicie niepolarne, może zawierać zarówno części polarne, jak i niepolarne i będzie zorientowane jak cząsteczka środka powierzchniowo czynnego.
W każdym przypadku podczas solubilizacji następuje wzrost masy micelarnej i liczby agregacji nie tylko na skutek włączenia solubilizatu, ale także na skutek wzrostu liczby cząsteczek surfaktantu niezbędnych do utrzymania stanu równowagi.
Solubilizacja jest tym skuteczniejsza, im niższa jest masa cząsteczkowa solubilizatu.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
Skuteczność surfaktantów zależy od wartości CMC.
Nacisk warstwy powierzchniowej 2D

→ -siły napięcia powierzchniowego.
- dwuwymiarowe ciśnienie.
Warstwa powierzchniowa to siła równa różnicy napięcia powierzchniowego roztworu środka powierzchniowo czynnego i czystego rozpuszczalnika, skierowana w stronę czystej powierzchni.
Pomiędzy roztworem a warstwą powierzchniową ustala się równowaga



Na  istnieje obszar, w którym
istnieje obszar, w którym  zależy liniowo od stężenia.
zależy liniowo od stężenia.
G [mol/m2].
 -powierzchnia zajmowana przez jeden mol substancji
-powierzchnia zajmowana przez jeden mol substancji
Wtedy dwuwymiarowa izoterma ciśnienia będzie miała postać 
 - dwuwymiarowa izoterma ciśnienia.
- dwuwymiarowa izoterma ciśnienia.
Uzależnienie  od SM:
od SM:

Na  - ciśnienie dwuwymiarowe gwałtownie wzrasta. Na
- ciśnienie dwuwymiarowe gwałtownie wzrasta. Na  dwuwymiarowość ulega deformacji, powodując nagły wzrost
dwuwymiarowość ulega deformacji, powodując nagły wzrost  .
.
Folię ograniczoną po obu stronach identycznymi fazami nazywa się dwustronną. W takich filmach obserwuje się ciągły ruch ługu macierzystego.
Folie o grubości mniejszej niż 5 nm nazywane są foliami czarnymi.

Warstwy adsorpcyjne muszą posiadać dwie cechy: lepkość i łatwość przemieszczania się, płynność i elastyczność.
Efekt Marangoni ma charakter samoleczenia.
Trójkąt Gibbsa,  - nadciśnienie.
- nadciśnienie.
Folia się rozciągnęła i w związku z opuszczeniem części cieczy, środki powierzchniowo czynne przedostają się do wolnej przestrzeni. Trójkąt Gibbsa.
Wpływ siły adsorpcji ciał.
Na powierzchni folii zawsze znajduje się warstwa adsorpcyjna, po co wtedy
Równanie Langmuira:


 w dwuwymiarowe ciśnienie
w dwuwymiarowe ciśnienie
 - analog równania Szyszkowskiego
- analog równania Szyszkowskiego
Zjawiska elektrokinetyczne. Elektryczna podwójna warstwa (EDL).
Model Gelemholtza. Teoria Gouya-Chapmana.
Lot 1808

U – rurkę kształtową, zanurz w niej 2 elektrody. Naruszone zostaje prawo naczyń połączonych i następuje zmiana poziomu cieczy w rurce – zjawiska elektrokinetyczne.
Zjawiska kinetyczne:
Elektroforeza
Elektroosmoza
Potencjał przepływu (przepływu).
Potencjał sedymentacyjny
1 i 2 powstają po przyłożeniu różnicy potencjałów, 3 i 4 przebijanie i sedymentacja cząstek koloidalnych powoduje pojawienie się różnicy potencjałów.
Elektroosmoza jest ruchem ośrodka dyspersyjnego względem stacjonarnej fazy rozproszonej pod wpływem prądu elektrycznego.
Elektroforeza – jest to ruch cząstek fazy rozproszonej względem stacjonarnego ośrodka dyspersyjnego pod wpływem prądu elektrycznego.
P  Przyczyną występowania zjawisk elektrokinetycznych jest przestrzenne rozdzielenie ładunków i pojawienie się podwójnej warstwy elektrycznej.
Przyczyną występowania zjawisk elektrokinetycznych jest przestrzenne rozdzielenie ładunków i pojawienie się podwójnej warstwy elektrycznej.
Podwójna warstwa elektryczna to płaski kondensator, jedna płytka jest utworzona przez jony określające potencjał, druga przez przeciwjony. Jony są zanieczyszczane w ten sam sposób, w jaki kojony determinujące potencjał są wypychane do objętości roztworu. Odległość między płytami  . Potencjał spada liniowo, różnica potencjałów
. Potencjał spada liniowo, różnica potencjałów  .
.
Zewnętrzna różnica potencjałów powoduje pojawienie się modułu sprężystości przy ścinaniu  jest parą sił na jednostkę powierzchni działających wzdłuż powierzchni ciała stałego.
jest parą sił na jednostkę powierzchni działających wzdłuż powierzchni ciała stałego.

W równowadze moduł ścinania jest równy modułowi tarcia lepkiego (  ).
).

W naszych warunkach  ,
,

 - Równanie Gelemholtza-Smalukowskiego
- Równanie Gelemholtza-Smalukowskiego
 - prędkość liniowa przesunięcia fazowego.
- prędkość liniowa przesunięcia fazowego.
mi– natężenie pola elektrycznego.
 - różnica potencjałów pomiędzy płytkami
- różnica potencjałów pomiędzy płytkami
 - ruchliwość elektroforetyczna [m 2 /(V*s)].
- ruchliwość elektroforetyczna [m 2 /(V*s)].
Model Helemholtza nie uwzględnia ruchu termicznego cząsteczek. W rzeczywistości rozkład jonów w warstwie podwójnej jest bardziej złożony.
Gui i Chapman zidentyfikowali następujące przyczyny DES:
Przejście jonu z jednej fazy do drugiej po ustaleniu równowagi.
Jonizacja materii fazy stałej.
Uzupełnianie powierzchni jonami obecnymi w ośrodku dyspersyjnym.
Polaryzacja z zewnętrznego źródła prądu.
Podwójna warstwa elektryczna ma strukturę rozmytą lub rozproszoną. Jony są zwykle równomiernie rozmieszczone w warstwie rozproszonej.
Warstwa rozproszona składa się z przeciwrynonów, których długość zależy od ich energii kinetycznej. W temperaturach bliskich zera absolutnego przeciwjony znajdują się jak najbliżej jonów determinujących potencjał.
Teoria Danyi opiera się na dwóch równaniach:
Równanie Boltzmanna

 - przeciwdziałać siłom oddziaływania elektrostatycznego.
- przeciwdziałać siłom oddziaływania elektrostatycznego.
 - objętościowa gęstość ładunku.
- objętościowa gęstość ładunku.

Równanie Poissona 
Ponieważ grubość EDL jest znacznie mniejsza niż wielkość cząstek, a dla płaskiego EDL jest to pochodna względem współrzędnych  I
I  zostaje zniesiony.
zostaje zniesiony. 
Dla e y na y<<1 функцию можно разложить в ряд Маклорена:

Ograniczmy się zatem do dwóch wyrazów szeregu:


 - Grubość DEL to odległość, o jaką maleje potencjał DEL mi raz.
- Grubość DEL to odległość, o jaką maleje potencjał DEL mi raz.
Im niższa temperatura, tym mniej  . W T → 0 – płaska DEL. Im wyższe stężenie, tym więcej I, tym mniej
. W T → 0 – płaska DEL. Im wyższe stężenie, tym więcej I, tym mniej  .
.





 „–” oznacza, że potencjał maleje wraz z odległością. =>
„–” oznacza, że potencjał maleje wraz z odległością. =>
=>

 ,
,
 - potencjał maleje wykładniczo.
- potencjał maleje wykładniczo.
Potencjał gęstości ładunku powierzchniowego: 
Ładunek powierzchniowy to ładunek objętościowy o przeciwnym znaku, całkowany po odległości.




=>


Gdzie potencjał zmniejsza się 2,7 razy - 
Pojemność podwójnej warstwy 
Wadą teorii jest to, że nie uwzględnia się obecności warstwy Helemholtza, tj. nie bierze pod uwagę  , stąd błędy w określeniu głównych parametrów. Nie wyjaśnia także wpływu jonów o różnym charakterze na grubość podwójnej warstwy elektrycznej.
, stąd błędy w określeniu głównych parametrów. Nie wyjaśnia także wpływu jonów o różnym charakterze na grubość podwójnej warstwy elektrycznej.
Teoria Sterna. Struktura miceli koloidalnej.
Podwójna warstwa elektryczna składa się z dwóch części: gęstej i rozproszonej. W wyniku oddziaływania jonów tworzących potencjał ze specyficznie zaadsorbowanymi jonami powstaje gęsta warstwa. Jony te z reguły są częściowo lub całkowicie odwodnione i mogą mieć taki sam lub przeciwny ładunek w stosunku do jonów determinujących potencjał. Zależy to od stosunku energii oddziaływania elektrostatycznego  i specyficzny potencjał adsorpcji
i specyficzny potencjał adsorpcji  . Jony gęstej warstwy są utrwalone. Pozostała część jonów znajduje się w warstwie rozproszonej, jony te są wolne i mogą przedostawać się głębiej do roztworu, tj. z obszaru o wyższym stężeniu do obszaru o niższym stężeniu. Całkowita gęstość ładunku składa się z dwóch części.
. Jony gęstej warstwy są utrwalone. Pozostała część jonów znajduje się w warstwie rozproszonej, jony te są wolne i mogą przedostawać się głębiej do roztworu, tj. z obszaru o wyższym stężeniu do obszaru o niższym stężeniu. Całkowita gęstość ładunku składa się z dwóch części.

 -ładunek warstwy Helmholtza
-ładunek warstwy Helmholtza
 -Ładunek warstwy rozproszonej
-Ładunek warstwy rozproszonej
Na powierzchni znajduje się pewna liczba centrów adsorpcji, z których każde oddziałuje z jednym przeciwjonem. Stała takiej reakcji quasi-chemicznej jest równa:

 , Gdzie
, Gdzie  - ułamek molowy przeciwjonów w roztworze
- ułamek molowy przeciwjonów w roztworze
Rozkład Helmholtza
Potencjał maleje liniowo
Rozkład potencjału Gouy’a. Nie ma gęstej warstwy, potencjał maleje wykładniczo od wartości 
Dystrybucja Sterna.
Początkowo potencjalny spadek ma charakter liniowy, a następnie wykładniczy.
Kiedy w przypadku elektroforezy przyłożone zostanie pole elektryczne, to nie cząstka fazy stałej porusza się bezpośrednio, ale cząstka fazy stałej otoczona warstwą jonów. DES powtarza kształt cząstki fazy rozproszonej. Po przyłożeniu potencjału część warstwy rozproszonej zostaje oderwana. Linia przerwania nazywa się przesuwająca się granica.
Potencjał powstający na granicy poślizgu w wyniku oddzielenia się części warstwy rozproszonej nazywa się potencjałem potencjał elektrokinetyczny(Potencjał zeta  ).
).
Nazywa się cząstką fazy rozproszonej z otaczającą warstwą przeciwjonów i podwójną warstwą elektryczną micela.
Zasady pisania miceli koloidalnych:


Elektrolit ładujący 1-1
T – cząstka fazy rozproszonej.
AA jest granicą pomiędzy częściami gęstymi i rozproszonymi.
BB – granica ruchoma.
Przesuwana granica może, ale nie musi, pokrywać się z linią AA.
Wartość pH, przy której potencjał zeta wynosi zero, nazywa się punkt izoelektryczny.
CaCl2 + Na2SO4 → CaSO4 ↓ + 2NaCl
1. Nadmiar CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 X + X Cl - - zapis miceli.
CaSO 4 m – kruszywo.
CaSO 4 m∙nCa 2+ – rdzeń.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - cząstka.
2. Nadmiar Na2SO4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micela
CaSO 4 m – kruszywo.
CaSO 4 m∙nSO 4 2 + – rdzeń.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - cząstka
Równanie Gelemholtza-Smoluchowskiego

 - prędkość liniowa przemieszczenia granicy (w elektroosmozie).
- prędkość liniowa przemieszczenia granicy (w elektroosmozie).
 - różnica potencjałów na płytkach kondensatora (w elektroosmozie).
- różnica potencjałów na płytkach kondensatora (w elektroosmozie).


 - objętościowe natężenie przepływu roztworu, S– pole przekroju poprzecznego komórki.
- objętościowe natężenie przepływu roztworu, S– pole przekroju poprzecznego komórki.

mi– natężenie pola elektrycznego.


 (na elektroosmozę).
(na elektroosmozę).
Dla potencjału przepływu:

 - potencjał
- potencjał
 - nacisk na membranę
- nacisk na membranę
Z reguły wartości ruchliwości elektroforetycznej i ruchliwości elektroosmotycznej są mniejsze niż obliczone. Dzieje się tak z powodu:
Efekt relaksacji (ruch cząstki fazy rozproszonej powoduje naruszenie symetrii atmosfery jonowej).
Hamowanie elektroforetyczne (pojawienie się dodatkowego tarcia w wyniku ruchu przeciwjonów).
Zniekształcenie linii prądu w przypadku cząstek przewodzących prąd elektryczny.
Zależność napięcia powierzchniowego od potencjału. Równanie Lippmanna.
Tworzenie się EDL następuje samoistnie w wyniku chęci układu do zmniejszenia jego energii powierzchniowej. W warunkach stałych T I P uogólnione równanie pierwszej i drugiej zasady termodynamiki wygląda następująco:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. równanie Lippmanna.
- 1. równanie Lippmanna.
 - gęstość ładunku powierzchniowego.
- gęstość ładunku powierzchniowego.
 - pojemność różnicowa.
- pojemność różnicowa.
 - 2. równanie Lippmanna.
- 2. równanie Lippmanna.
Z- pojemność.
Rozwiążmy pierwsze równanie Lippmanna i podstawowe równanie adsorpcji:
 ,
,


 , Następnie
, Następnie
 - Równanie Nernsta
- Równanie Nernsta
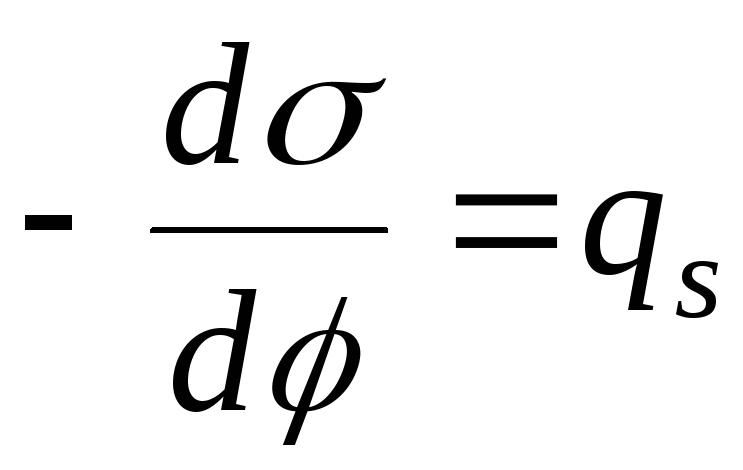 ,
,
 ,
,




 - równanie krzywej elektrokapilarnej (ECC).
- równanie krzywej elektrokapilarnej (ECC).
W  :
: , Ale
, Ale 
Kationowe środki powierzchniowo czynne (CPAS) redukują katodową gałąź EKC.
Anionowe środki powierzchniowo czynne (APS) redukują anodową gałąź EKC.
Niejonowe środki powierzchniowo czynne (NSAS) zmniejszają środkową część ECC.
Stabilność układów rozproszonych. Rozłączające ciśnienie.
Systemy rozproszone można podzielić:
Układy niestabilne termodynamicznie mogą być stabilne kinetycznie w wyniku przejścia do stanu metastabilnego.
Istnieją dwa rodzaje stabilności:
Stabilność sedymentacji (w stosunku do grawitacji).
Stabilność agregacyjna. (w odniesieniu do przyczepności)
Koagulacja to proces adhezji cząstek, prowadzący do utraty stabilności agregacji. Koagulacja może być spowodowana zmianami temperatury, pH, mieszaniem i ultradźwiękami.
Wyróżnia się koagulację:
Odwracalny.
Nieodwracalny.
Koagulacja następuje po wprowadzeniu elektrolitów.
Zasady krzepnięcia:

Film- jest to część układu zlokalizowana pomiędzy dwiema powierzchniami międzyfazowymi.
Rozłączające ciśnienie występuje, gdy grubość folii gwałtownie maleje w wyniku interakcji zbliżających się warstw powierzchniowych.

„-” - w miarę zmniejszania się grubości warstwy wzrasta ciśnienie rozłączające.
P 0 to ciśnienie w fazie objętościowej, która jest kontynuacją międzywarstwy.
P 1 – ciśnienie w folii.
Teoria stabilności. DLFO (Deryagin, Landau, Fairway, Overbeck).
Zgodnie z teorią DLFO ciśnienie rozłączające składa się z dwóch składników:
Elektrostatyczny PE (dodatni, wynika to z sił odpychania elektrostatycznego). Odpowiada spadkowi energii Gibbsa wraz ze wzrostem grubości warstwy.
Molekularny P M (ujemny, ze względu na działanie sił przyciągających). Jest to spowodowane kompresją folii pod wpływem chemicznych sił powierzchniowych, promień działania sił wynosi dziesiąte części nm przy energii około 400 kJ/mol.
Całkowita energia interakcji:

 - system jest agregatywnie stabilny
- system jest agregatywnie stabilny
 - niestabilny system
- niestabilny system
P  składnik pozytywny.
składnik pozytywny.
Wzrost wynika ze wzrostu energii potencjalnej podczas ściskania cienkich folii. W przypadku folii o dużej grubości nadmiar energii jonów jest kompensowany i jest równy oddziaływaniu energii w objętości ośrodka dyspersyjnego.
Jeśli  (
( - grubość folii,
- grubość folii,  - promień jonu) rozrzedzenie filmu prowadzi do zaniku i redukcji cząsteczek i jonów przy minimalnej energii powierzchniowej w nim zawartej. Zmniejsza się liczba sąsiadujących cząstek, w wyniku czego wzrasta energia potencjalna cząstek pozostających w folii.
- promień jonu) rozrzedzenie filmu prowadzi do zaniku i redukcji cząsteczek i jonów przy minimalnej energii powierzchniowej w nim zawartej. Zmniejsza się liczba sąsiadujących cząstek, w wyniku czego wzrasta energia potencjalna cząstek pozostających w folii.


Teoria DLVO traktuje oddziaływanie cząstek jako oddziaływanie płytek.

Cząsteczki nie oddziałują



 - równanie Laplace'a,
- równanie Laplace'a,  ,
,


Do słabo naładowanych powierzchni
W przypadku silnie naładowanych powierzchni:

Składnik molekularny to oddziaływanie dwóch atomów:
 ~
~
Oddziaływanie atomu z powierzchnią:

Weźmy dwa rekordy:
D  Aby otrzymać składnik molekularny, należy zsumować wszystkie energie interakcji atomów prawej i lewej płytki.
Aby otrzymać składnik molekularny, należy zsumować wszystkie energie interakcji atomów prawej i lewej płytki.
Gdzie  - Stała Hamakera (uwzględnia naturę oddziałujących ciał).
- Stała Hamakera (uwzględnia naturę oddziałujących ciał).
To. energię interakcji cząstek w układzie można wyrazić za pomocą krzywych potencjału.
I – minimum potencjału pierwotnego. Jest to strefa nieodwracalnej koagulacji, w której przeważają siły przyciągania.
II – strefa stabilności agregatowej, w której dominują siły odpychające.
III – minimum potencjału wtórnego (lub strefa flokulacji). Pomiędzy cząstkami fazy rozproszonej znajduje się warstwa elektrolitu, którą cząstki można oddzielić i przenieść do strefy stabilności agregacji.

Krzywa 1 – system jest agregatywnie stabilny.
Krzywa 2 – stabilna w strefie I, niestabilna w strefie II.
Krzywa 3 – w układzie nastąpiła koagulacja.
Krzywa 4 – w punkcie 4 całkowita energia oddziaływania U=0,  , ten ekstremalny punkt odpowiada początkowi szybkiej koagulacji.
, ten ekstremalny punkt odpowiada początkowi szybkiej koagulacji.
Istnieją dwa przypadki:
1. Lekko naładowane powierzchnie:
U = U mi + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - jest to grubość warstwy odpowiadająca początkowi procesu koagulacji.
- jest to grubość warstwy odpowiadająca początkowi procesu koagulacji.






 - do słabo naładowanych powierzchni
- do słabo naładowanych powierzchni
 Następnie
Następnie 
2. W przypadku silnie naładowanych powierzchni:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Ustawmy kwadrat (3)

Koagulacja:
W przypadku adsorpcji specyficznej jony mogą być adsorbowane w ilościach superrównoważnych, tak że powierzchnia może zmieniać swój ładunek. Powierzchnia jest ponownie naładowana.
W przypadku adsorpcji specyficznej adsorbowane mogą być nie tylko jony o znakach przeciwnych, ale także o tym samym znaku.
Jeśli zaadsorbowane zostaną jony o tym samym znaku co powierzchnia, to w warstwie powierzchniowej nie nastąpi spadek potencjału, ale jego wzrost.

Koagulacja neutralizująca (zachodzi przy udziale cząstek słabo naładowanych i zależy nie tylko od ładunku koagulatora elektrolitowego, ale także od potencjału na granicy warstw gęstej i rozproszonej).
Teoria szybkiej koagulacji Smoluchowskiego.

Zależność szybkości krzepnięcia od stężenia elektrolitu.
I – szybkość krzepnięcia jest niska,
II – szybkość koagulacji jest prawie proporcjonalna do stężenia elektrolitu.
III – obszar szybkiej koagulacji, prędkość jest praktycznie niezależna od stężenia.
Podstawowe postanowienia:
Początkowy zol jest monodyspersyjny, podobne cząstki mają kształt kulisty.
Wszystkie zderzenia cząstek są skuteczne.
Kiedy zderzają się dwie cząstki pierwotne, powstaje cząstka wtórna. Szkoła średnia + podstawowa = trzeciorzędna. Pierwotne, wtórne, trzeciorzędne – wielość.
Pod względem kinetyki chemicznej proces koagulacji można opisać równaniem:
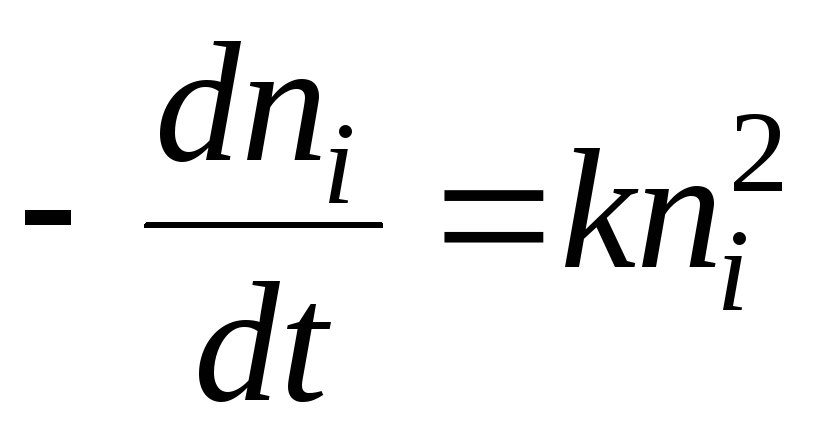
Rozwiązaniem będzie równanie: 
 - połowa czasu krzepnięcia. Jest to czas, w którym liczba cząstek zolu zmniejsza się 2-krotnie.
- połowa czasu krzepnięcia. Jest to czas, w którym liczba cząstek zolu zmniejsza się 2-krotnie.

 ,
,
 ,
,
 ,
,


Wraz ze wzrostem krotności maksimum krzywych koagulacji przesuwa się w stronę większych wartości  .
.
Wady:
Założenie monodyspersyjności.
Założenie o skuteczności wszystkich zderzeń.


