4.D, L - System oznaczania stereoizomerów.
W wielu przypadkach do wyznaczenia konfiguracji absolutnej korzystne jest stosowanie nie systemu R,S, lecz innego systemu D,L. Wybór ekspresji izomeru D lub L opiera się na konkretnej lokalizacji. grupa reger w projekcji Fischera . D ,L-nomenklatura jest szeroko stosowana w nazwach -amino, -hydroksykwasy i węglowodany.
Według tego systemu konfiguracja L jest przypisana do stereosomeru, w którym w projekcji Fischera grupa odniesienia znajduje się na lewo od linii pionowej (od łacińskiego „laevus” - po lewej). W związku z tym, jeśli grupa odniesienia znajdujący się w projekcji Fischera po prawej stronie, stereoizomer ma konfigurację D (od łacińskiego „dexter” - po prawej):

Oczywiście musimy pamiętać, że w projekcji Fischera najbardziej utleniony atom węgla znajduje się na górze (czyli grupa COOH w kwasy amino- i hydroksylowe oraz grupa CH=O w węglowodanach).
Amino i hydroksykwasy
W -amino- i -hydroksykwasach grupami odniesienia są odpowiednio grupy NH2 i OH:

Jeśli aminokwas lub hydroksykwas zawiera kilka grup aminowych lub hydroksylowych, należy wskazać ich względną pozycję za pomocą przedrostków „erythro”, „treo” itp. O przynależności kwasu do serii D lub L decyduje grupa NH2 lub OH znajdująca się w pozycji - do grupy COOH znajdującej się u góry w rzucie Fischera:

W tym przypadku litery D i L, wskazujące pozycję grupy odniesienia, są oznaczone indeksem „S”. Ma to na celu uniknięcie nieporozumień. Indeks „S” podkreśla, że wskazana jest konfiguracja górnego centrum chiralnego, które znajduje się w pozycji - w stosunku do grupy karboksylowej, jak w aminokwasie serynie („S” - od słowa „seryna”).

W przypadku hydroksykwasów z kilkoma grupami OH, a także aminohydroksykwasów stosuje się alternatywne oznaczenie konfiguracji, w którym grupą odniesienia jest najniższa grupa HO w rzucie Fischera. W tym przypadku deskryptory konfiguracji D i L są oznaczone indeksem dolnym „g” (od „aldehyd glicerynowy”). W tym przypadku aminokwasy pokazane na ryc. 123 i 124 nazywane są: D g -treonina (L s - treonina) i L g -treonina ( D s - treonina).
Węglowodany.
W przypadku węglowodanów grupą odniesienia są: najniższy w projekcji Fischera grupa hydroksylowa związana z ammetycznym atomem węgla


Oczywiście w przypadku cząsteczek z jednym atomem asymetrycznym nazewnictwo D,L, podobnie jak nazewnictwo R,S, jednoznacznie wskazuje na absolutną konfigurację centrum chiralności. To samo dotyczy użycia D, L - nazwy stereoizomerów z kilkoma atomami asymetrycznymi, ponieważ w tym przypadku konfigurację pozostałych centrów chiralności podaje się przedrostkami erytro-, treo-, rybo-, lyxo- itp. Jeśli więc powiemy „threose”, to tylko ustawimy względny konfiguracja asymetrycznych atomów w cząsteczce. Wtedy nie będzie jasne, o którym enancjomerze mówimy: (26) czy (27), Jeśli powiemy „D-treoza”, to jednoznacznie wskażemy, że mamy na myśli izomer (26), ponieważ w nim odniesienie OH grupa znajduje się po prawej stronie w rzucie Fischera:

Zatem nazwa „D-treoza” (podobnie jak „L-treoza”) odnosi się do absolutnej konfiguracji obu asymetrycznych atomów w cząsteczce.
Podobnie jak nomenklatura R, S, system nazewnictwa stereoizomerów D, L nie jest powiązany ze znakiem skręcalności optycznej.
Należy zaznaczyć, że dotychczas małymi literami d określano kierunek obrotu płaszczyzny polaryzacji światła
(po prawej) i l
(lewy). Nie należy mylić użycia tych liter z użyciem wielkich liter D
i L, aby wskazać konfigurację cząsteczek. Obecnie kierunek obrotu płaszczyzny polaryzacji światła oznacza się zwykle symbolami (+) i (-).
5. Cząsteczki chiralne bez atomów asymetrycznych
W poprzednich rozdziałach rozważaliśmy cząsteczki, których chiralność jest zdeterminowana pewnym przestrzennym rozmieszczeniem czterech różnych atomów lub grup atomów względem pewnego centrum, zwanego centrum chiralności.
Mogą zdarzyć się przypadki, gdy w cząsteczce nie ma takich centrów, ale mimo to cząsteczka jest chiralna, ponieważ brakuje jej elementów symetrii grupy Sn. W takich przypadkach enancjomery różnią się rozmieszczeniem atomów
względem jakiejś osi lub płaszczyzny, zwanej osią chiralną lub płaszczyzną chiralną. Oś chiralności występuje na przykład w cząsteczkach kumulenu.
Budowa cząsteczki najprostszego kumulenu – allenu – jest taka, że jej dwa fragmenty CH 2 znajdują się w dwóch wzajemnie prostopadłych płaszczyznach:

Cząsteczka allenu jest achiralna: ma dwie płaszczyzny symetrii (pokazane na rysunku). Cząsteczki butadienu-1,2 i 3-metylo-butadienu-1,2 są również achiralne

Jeśli spojrzymy na cząsteczkę pentadienu-2,3, zobaczymy, że nie ma ona płaszczyzn symetrii (tak jak nie ma innych elementów symetrii grupy Sn). Ten dien występuje w postaci pary enancjomerów:

Chiralność cząsteczek (28) i (29) wynika z pewnego przestrzennego ułożenia podstawników względem osi (pokazanej na rysunku) przechodzącej przez atomy węgla połączone wiązaniami podwójnymi. Ta oś nazywa się oś chiralności. Mówi się, że cząsteczki takie jak (28) i (29) mają chiralność osiową.
Osie chiralności występują także w cząsteczkach niektórych innych związków, np. związków spiro (spiranów):

Wspomniane antropoizomery orto-dipodstawionych bifenyli są także cząsteczkami o chiralności osiowej.Przykłady cząsteczek o płaszczyzna chiralności cząsteczki para-cyklofanów mogą służyć:

Przedstawione tutaj enancjomery nie mogą przekształcać się w siebie w wyniku rotacji wokół wiązań - ze względu na wymagania przestrzenne fragmentów zawartych w tych cząsteczkach.
Nomenklaturę R, S można zastosować do określenia konfiguracji cząsteczek o chiralności osiowej i planarnej. Zainteresowani mogą znaleźć opis zasad przypisywania konfiguracji R lub S takim cząsteczkom w publikacji VINITI: IUPAC Nomenclature Rules for Chemistry, vol. 3, semi-volume 2, M., 1983.
6. Do reguły kolejności w R, S - nomenklatura.
W wielu przypadkach przy ustalaniu kolejności pierwszeństwa posłów pojawiają się komplikacje.Rozważmy niektóre z nich.
Przykład 1.
Jest oczywiste, że w tym przypadku młodszymi podstawnikami przy asymetrycznym atomie węgla, oznaczonymi gwiazdką, są H (d) i CH3 (c). Rozważmy dwa pozostałe złożone podstawniki, układając ich atomy warstwami.

W pierwszej warstwie obu podstawników atomy są identyczne. W drugiej warstwie zestaw atomów jest również taki sam. (H, C, O). Dlatego musimy zwrócić się do trzeciej warstwy atomów. W takim przypadku należy przede wszystkim porównać lewy i prawy podstawnik atomy warstwy III powiązane ze starszymi atomami warstwy II(to znaczy, należy wziąć pod uwagę „starsze oddziały” obu pełnomocników). W tym przypadku mówimy o atomach związanych z atomem tlenu warstwy P. Ponieważ atom C w prawym podstawniku jest związany z atomem tlenu, a atom H jest związany z atomem tlenu w lewym podstawniku, prawy podstawnik ma pierwszeństwo:

Do połączenia należy przypisać konfigurację R:

Gdyby atomy „starszej gałęzi” w trzeciej warstwie okazały się takie same, na przykład oba C, wówczas konieczne byłoby porównanie atomów tej samej III warstwy, ale w młodszej gałęzi. Wtedy lewy zastępca zyskałby przewagę. Jednak w naszych porównaniach nie dochodzimy do tego punktu, ponieważ możemy dokonać wyboru na podstawie różnic w atomach warstwa starszej gałęzi.
W zupełnie podobny sposób wyboru kolejności pierwszeństwa dokonuje się na przykład pomiędzy następującymi podstawnikami:

Może zaistnieć sytuacja, gdy w celu wyłonienia starszego zastępcy konieczne będzie „przejście” przez połączenie wielokrotne. W takim przypadku warto skorzystać z pomocy tzw atomy fantomowe, mający zerową liczbę atomową (to znaczy a priori najniższą) i wartościowość równą 1.

W tym przykładzie nadr dokonuje wyboru pomiędzy lewoskrętnymi i prawoskrętnymi podstawnikami zawierającymi węgiel. Rozważmy je, uprzednio „otwierając” podwójne wiązanie C=C pierwszego podstawnika. W takim przypadku pojawią się zduplikowane atomy (zaznaczone w kółkach). Do zduplikowanych atomów dodamy atomy fantomowe (oznaczmy je literą f), aby wartościowość każdego z nich wynosiła 4:

Teraz możemy porównać lewy i prawy podstawnik:

Różnica w trzeciej warstwie atomów pozwala dać pierwszeństwo prawemu podstawnikowi:

Dlatego połączenie ma konfigurację R.
Przykład 3. W niektórych przypadkach dwa podstawniki na atomie asymetrycznym są strukturalnie identyczne, ale różnią się jedynie konfiguracją absolutną centrów chiralnych. Potem to akceptują – Konfiguracja R jest starsza niż konfiguracja S. W związku z tym centralnemu atomowi węgla w poniższym przykładzie należy przypisać konfigurację S:

Przykład 4. Zasady przedstawione powyżej mają zastosowanie również do opisu konfiguracji absolutnej asymetrycznych atomów z trzema podstawnikami (atomy azotu, fosforu, siarki). W tym przypadku jako czwarty podstawnik, który jest zawsze najmłodszy, stosuje się atom fantomowy (za atom fantomowy można uznać samotną parę elektronów):

Przykład 5. Czasami, aby wybrać starszeństwo podstawników, konieczne jest „otwarcie” cyklu, tak jak dokonuje się „otwarcia” wiązania wielokrotnego.

W tym przypadku łatwo jest wyznaczyć najstarszy (O) i najmłodszy (H) podstawnik przy atomie węgla oznaczonym gwiazdką. Aby dokonać wyboru pomiędzy atomami węgla 1 Cu 2 C należy „otworzyć” cykl na wiązaniu 2 C-O według poniższego schematu (w kółkach zaznaczono dwójki atomów):

W tym przypadku, w przeciwieństwie do „otwarcia” wiązań wielokrotnych, zduplikowane atomy nie reprezentują już gałęzi „ślepych zaułków”, ale kontynuują powtarzanie atomu oznaczonego gwiazdką. Oznacza to, że procedura „otwarcia” cyklu kończy się w momencie, gdy na końcach obu gałęzi pojawi się ten sam atom (a raczej jego duplikat). Teraz możemy porównać atomy 1 C 2 C, biorąc pod uwagę odpowiednie warstwy atomów:

Różnica w trzeciej warstwie pozwala na przewagę stażu - atom węgla 2 C. W związku z tym dane centrum chiralne ma konfigurację S:

1.E.Iliel, Podstawy stereochemii. M.: Mir, 1971, 107 s.
2.V.M.Potapow, Stereochemia. M.: Chemia, 1988, 463 s.
3.V.I.Sokolov, Wprowadzenie do stereochemii teoretycznej, M., Nauka, 1979, 243 s.
Aby określić konfigurację absolutną centrum chiralnego, należy wykonać następujące operacje:
1. Ustaw centrum chiralne tak, aby linia wzroku była skierowana od chiralnego węgla do młodszego podstawnika.
2. W wynikowym rzucie trzy pozostałe podstawniki będą umieszczone pod kątem 120 o. Jeśli starszeństwo podstawników maleje zgodnie ze wskazówkami zegara- Ten R-konfiguracja (zakłada się następujące zmiany pierwszeństwa: A > D > B):
Jeśli przeciwnie do ruchu wskazówek zegara - S-konfiguracja:
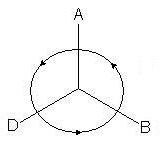
Konfigurację absolutną można wyznaczyć za pomocą wzoru Fishera. Aby to zrobić, poprzez działania, które nie zmieniają wzoru Fischera, umieszcza się młodszy podstawnik. Następnie rozważa się zmianę stażu pracy trzech pozostałych zastępców. Jeśli starszeństwo podstawników maleje zgodnie z ruchem wskazówek zegara, jest to konfiguracja R; jeśli maleje w przeciwnym kierunku, jest to konfiguracja S. Młodszy zastępca nie jest brany pod uwagę.
Przykład
Rozważmy określenie konfiguracji centrów chiralnych na przykładzie 3-bromo-2-metylo-2-chlorobutanolu-1, który ma następującą budowę:
Ustalmy konfigurację absolutną C2. Aby to zrobić, wyobraźmy sobie C 3 i C 4, a także wszystko, co z nimi związane, w postaci rodnika A:

Teraz oryginalna formuła będzie wyglądać następująco:
Określamy starszeństwo podstawników (od starszego do młodszego): Cl > A > CH 2 OH > CH 3 . Dokonujemy parzystej liczby przegrupowań (nie zmienia to stereochemicznego znaczenia wzoru!), tak aby najniższy podstawnik znalazł się na dole:

Rozważmy teraz trzy górne podstawniki we wzorze Fischera w centrum chiralności C2:
Można zauważyć, że przejście tych podstawników w kolejności malejącej starszeństwa następuje w kierunku przeciwnym do ruchu wskazówek zegara, stąd konfiguracja tego centrum chiralnego to S.
Podobne działania wykonamy dla innego centrum chiralnego związanego z C3. Wyobraźmy sobie jeszcze raz, tym razem C2 i wszystko z nim związane, w postaci rodnika W:

Teraz oryginalna formuła będzie wyglądać następująco:
Ponownie określamy starszeństwo podstawników (od starszego do młodszego): Br > B > CH 3 > H. Dokonujemy parzystej liczby permutacji tak, aby najmłodszy podstawnik znów znalazł się na dole:

Ustalmy, w którą stronę następuje spadek stażu pracy (nie bierzemy pod uwagę najniższego, najmłodszego posła!):
Spadek starszeństwa podstawników następuje w kierunku przeciwnym do ruchu wskazówek zegara, dlatego konfiguracja tego centrum chiralnego to S.
Nazwa substancji wyjściowej z uwzględnieniem konfiguracji absolutnej centrów chiralnych - 3-/S/-bromo-2-/S/-metylo-2-chlorobutanol-1
Jak oznaczyć konfigurację związku, aby nazwa odzwierciedlała przestrzenny układ grup przy chiralnym atomie węgla? Do tego używają R, S-system zaproponowany przez K. Ingolda, R. Kahna, Z. Preloga. R, S-system opiera się na określeniu starszeństwa podstawników wokół centrum chiralnego. Staż grupowy ustala się w następujący sposób:
1). Atom o wyższej liczbie atomowej jest starszy od atomu o niższej liczbie atomowej.
2). Jeżeli atomy bezpośrednio połączone z węglem C* są takie same, wówczas należy wziąć pod uwagę starszeństwo kolejnych atomów.
Na przykład, jak określić najstarszą z grup: -C 2 H 5 i CH (CH 3) 2 w związku

W grupie etylowej po atomie połączonym z centrum chiralnym następują H, H i C, a w grupie izopropylowej - H, C i C. Porównując te grupy między sobą ustalamy, że grupa izopropylowa jest starsza od grupa etylowa.

3). Jeśli chiralny węgiel C* jest połączony z atomem mającym wiązanie wielokrotne, wówczas wiązania tego atomu należy przedstawić jako wiązania proste.

4). W celu ustalenia konfiguracji cząsteczki ustawia się ją tak, aby wiązanie centrum chiralnego z grupą podrzędną nr 4 było skierowane od obserwatora i określa się położenie pozostałych grup (ryc. 2.6).

Ryż. 2.6. Definicja R, S-konfiguracje
Jeśli starszeństwo grup maleje (1®2®3) zgodnie z ruchem wskazówek zegara, wówczas konfigurację centrum chiralnego określa się jako R(od łacińskiego słowa „rectus” - po prawej). Jeśli starszeństwo podstawników maleje w kierunku przeciwnym do ruchu wskazówek zegara, wówczas konfiguracja centrum chiralnego jest taka S(od łacińskiego „złowrogiego” - po lewej).
Znak skręcalności optycznej (+) lub (-) wyznacza się eksperymentalnie i nie ma on związku z oznaczeniem konfiguracji ( R) Lub ( S). Na przykład prawoskrętny 2-butanol ma ( S)-konfiguracja.
Aby określić konfigurację połączenia przedstawioną wzorem rzutowania Fischera, należy postępować w następujący sposób.

1). Wykonaj parzystą liczbę permutacji podstawników w centrum chiralności (nieparzysta liczba permutacji doprowadzi do enancjomeru), tak aby najniższy podstawnik nr 4 znajdował się na górze lub na dole.

2). Określ lokalizację pozostałych grup, przeglądając je w kolejności malejącego stażu pracy. Jeśli starszeństwo podstawników maleje zgodnie z ruchem wskazówek zegara, wówczas początkową konfigurację określa się jako R-konfiguracja, jeśli przeciwnie do ruchu wskazówek zegara, wówczas konfiguracja jest zdefiniowana jako S-konfiguracja.
Jeśli przekształcenie wzoru projekcji nie jest łatwe, możesz ustalić kolejność malejącego pierwszeństwa, odrzucając młodszy podstawnik stojący z boku, ale wybierz symbol „odwrotny” do oznaczenia konfiguracji. Na przykład w oryginalnym połączeniu

odrzucając młodszego zastępcę (H), ustalamy kolejność malejącego stażu pracy: 1 → 2 → 3. Otrzymujemy oznaczenie ( S), zmień go na ( R) i uzyskaj poprawną nazwę: ( R kwas )-2-chloroetanosulfonowy.
Aby uruchomić tę aplikację, musisz włączyć JavaScript.Konfiguracja elektronowa atomu to wzór pokazujący rozmieszczenie elektronów w atomie według poziomów i podpoziomów. Po przestudiowaniu artykułu dowiesz się, gdzie i jak znajdują się elektrony, zapoznasz się z liczbami kwantowymi i potrafisz zbudować konfigurację elektronową atomu według jego liczby, na końcu artykułu znajduje się tabela pierwiastków.
Po co badać konfigurację elektroniczną elementów?
Atomy są jak zbiór konstrukcyjny: jest pewna liczba części, różnią się one od siebie, ale dwie części tego samego typu są absolutnie takie same. Ale ten zestaw konstrukcyjny jest o wiele ciekawszy niż plastikowy i oto dlaczego. Konfiguracja zmienia się w zależności od tego, kto jest w pobliżu. Na przykład tlen obok wodoru Może zamienia się w wodę, w pobliżu sodu zamienia się w gaz, a w pobliżu żelaza całkowicie zamienia się w rdzę. Aby odpowiedzieć na pytanie, dlaczego tak się dzieje i przewidzieć zachowanie atomu obok drugiego, konieczne jest zbadanie konfiguracji elektronowej, co zostanie omówione poniżej.
Ile elektronów jest w atomie?
Atom składa się z jądra i krążących wokół niego elektronów; jądro składa się z protonów i neutronów. W stanie neutralnym każdy atom ma liczbę elektronów równą liczbie protonów w jego jądrze. Liczbę protonów wyznacza się liczbą atomową pierwiastka, np. siarka ma 16 protonów – jest to 16. element układu okresowego. Złoto ma 79 protonów – jest to 79. element układu okresowego. Odpowiednio siarka ma 16 elektronów w stanie neutralnym, a złoto ma 79 elektronów.
Gdzie szukać elektronu?
Obserwując zachowanie elektronu wyprowadzono pewne wzorce, które opisuje się liczbami kwantowymi, jest ich w sumie cztery:
- Główna liczba kwantowa
- Orbitalna liczba kwantowa
- Magnetyczna liczba kwantowa
- Spinowa liczba kwantowa
Orbitalny
Ponadto zamiast słowa orbita użyjemy terminu „orbita”; orbital to funkcja falowa elektronu; z grubsza jest to obszar, w którym elektron spędza 90% swojego czasu.
N - poziom
L - skorupa
M l - liczba orbitalna
M s - pierwszy lub drugi elektron na orbicie
Orbitalna liczba kwantowa l
W wyniku badania chmury elektronów odkryli, że w zależności od poziomu energii chmura przybiera cztery główne formy: piłkę, hantle i dwie inne, bardziej złożone. W kolejności rosnącej energii formy te nazywane są powłokami s, p, d i f. Każda z tych powłok może mieć 1 (na s), 3 (na p), 5 (na d) i 7 (na f) orbitali. Orbitalna liczba kwantowa to powłoka, w której znajdują się orbitale. Orbitalna liczba kwantowa dla orbitali s, p, d i f przyjmuje odpowiednio wartości 0,1,2 lub 3.
Na powłoce s znajduje się jeden orbital (L=0) - dwa elektrony
Na powłoce p znajdują się trzy orbitale (L=1) - sześć elektronów
Na powłoce d znajduje się pięć orbitali (L=2) - dziesięć elektronów
Na powłoce f znajduje się siedem orbitali (L=3) - czternaście elektronów
Magnetyczna liczba kwantowa m l
Na powłoce p znajdują się trzy orbitale, są one oznaczone liczbami od -L do +L, czyli dla powłoki p (L=1) są orbitale „-1”, „0” i „1” . Magnetyczna liczba kwantowa jest oznaczona literą m l.
Wewnątrz powłoki elektrony łatwiej jest zlokalizować na różnych orbitaliach, dlatego pierwsze elektrony zapełniają po jednym na każdym orbicie, a następnie do każdego dodawana jest para elektronów.
Rozważ powłokę d:
Powłoka d odpowiada wartości L=2, czyli pięciu orbitali (-2,-1,0,1 i 2), pierwsze pięć elektronów wypełnia powłokę przyjmując wartości M l =-2, M l =-1, M l =0, M l =1, M l =2.
Spinowa liczba kwantowa m s
Spin to kierunek obrotu elektronu wokół własnej osi, są dwa kierunki, więc spinowa liczba kwantowa ma dwie wartości: +1/2 i -1/2. Jeden podpoziom energii może zawierać tylko dwa elektrony o przeciwnych spinach. Spinowa liczba kwantowa jest oznaczana m s
Główna liczba kwantowa n
Główną liczbą kwantową jest poziom energii, obecnie znanych jest siedem poziomów energii, każdy oznaczony cyfrą arabską: 1,2,3,...7. Liczba pocisków na każdym poziomie jest równa numerowi poziomu: na pierwszym poziomie znajduje się jeden pocisk, na drugim dwa itd.
Liczba elektronów

Zatem każdy elektron można opisać czterema liczbami kwantowymi, kombinacja tych liczb jest unikalna dla każdej pozycji elektronu, weźmy pierwszy elektron, najniższy poziom energii to N = 1, na pierwszym poziomie jest jedna powłoka, pierwsza skorupa na dowolnym poziomie ma kształt kuli (s -skorupy), tj. L=0, magnetyczna liczba kwantowa może przyjąć tylko jedną wartość, M l =0, a spin będzie równy +1/2. Jeśli weźmiemy piąty elektron (w jakimkolwiek atomie), to głównymi liczbami kwantowymi dla niego będą: N=2, L=1, M=-1, spin 1/2.
Pojawia się następujący problem; Jak w jakiś prostszy i wygodniejszy sposób wyznaczyć pewną konfigurację, aby nie rysować za każdym razem jej struktury? W tym celu najczęściej używany
symbole Zapis ten zaproponowali Kahn (Chemical Society, Londyn), K. Ingold (University College, Londyn) i V. Prelog (ETH, Zurich).
Zgodnie z tym systemem pierwszeństwo lub sekwencja podstawników, tj. czterech atomów lub grup związanych z asymetrycznym atomem węgla, jest najpierw określana w oparciu o regułę pierwszeństwa (sekcja 3.16).
Na przykład w przypadku asymetrycznego atomu węgla połączone są cztery różne atomy, a ich staż pracy zależy tylko od liczby atomowej, a im wyższa liczba atomowa, tym starszy jest podstawnik. Zatem, w malejącej kolejności pierwszeństwa, atomy są ułożone w następującej kolejności:

Następnie cząsteczkę ustawia się tak, aby najniższa grupa była skierowana od obserwatora i rozważa się położenie pozostałych grup. Jeśli staż pracy tych grup maleje zgodnie z ruchem wskazówek zegara, wówczas konfigurację oznacza się symbolem R (od łacińskiego rectus - po prawej); jeśli starszeństwo tych grup maleje w kierunku przeciwnym do ruchu wskazówek zegara, wówczas konfiguracja jest oznaczona symbolem (od łacińskiego złowrogiego - po lewej).
Zatem konfiguracje I i II wyglądają następująco:

i są odpowiednio oznaczone symbolami
Pełna nazwa związku optycznie czynnego odzwierciedla zarówno konfigurację, jak i kierunek rotacji, np. odmianę racemiczną można oznaczyć symbolem np. -chlorku sec-butylu.
(Oznaczanie związków z wieloma asymetrycznymi atomami węgla omówiono w Sekcji 3.17.)
Oczywiście nie należy mylić kierunku skręcalności optycznej związku (ta sama właściwość fizyczna prawdziwej substancji, co temperatura wrzenia lub topnienia) z kierunkiem naszego spojrzenia, gdy mentalnie układamy cząsteczkę w jakiś określony warunkowy sposób. Dopóki dla konkretnego związku nie zostanie ustalone eksperymentalnie powiązanie między konfiguracją a znakiem obrotu, nie można stwierdzić, czy znak odpowiada lub odpowiada -konfiguracji.


