An der Grenzfläche findet Adsorption statt. Daher ist es sinnvoll, die thermodynamische Beschreibung von Oberflächenphänomenen als Sonderfall der Thermodynamik heterogener Systeme zu betrachten.
Reis. 3.4. Gibbs-Adsorption: 1- Zweiphasen-Vergleichssystem, 2- echtes Zweiphasensystem mit einem ungleichmäßigen Bereich
In der Thermodynamik heterogener Systeme wird es verwendet Additivitätsprinzip Das ist wie folgt: Alle umfangreichen Eigenschaften eines heterogenen Systems sind gleich der Summe der entsprechenden umfangreichen Eigenschaften, die die Phasen gehabt hätten, bevor sie in Kontakt gebracht wurden. Bezeichnen wir die Phasen mit α und β (Abb. 4). Dann gelten für ein ideales System, bei dem die Eigenschaften der Phasen in der Nähe der Grenzfläche mit ihren Masseneigenschaften übereinstimmen, die folgenden Beziehungen für die innere Energie U, das Volumen V, die Masse (Anzahl der Mol) n und die Entropie S nach Herstellung des Gleichgewichts in ein heterogenes System:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Dies setzt voraus, dass Temperatur und Druck in beiden Phasen gleich sind.
Bei realen heterogenen Systemen trägt der Übergangsbereich an der Grenze zweier Phasen zusätzlich zu den umfangreichen Eigenschaften des Systems bei. Wenn Oberflächenphänomene auftreten, sollte man den Unterschied zwischen den umfangreichen Eigenschaften eines realen heterogenen Systems und den umfangreichen Eigenschaften eines Modellsystems berücksichtigen, in dem Oberflächenphänomene fehlen. Ein solches System wird als Vergleichssystem bezeichnet. Das Vergleichssystem hat die gleichen intensiven Parameter (T, P, C i ...) und das gleiche Volumen V wie das reale System (Abb. 4).
Unter dem Adsorptionswert G versteht man aus thermodynamischer Sicht die überschüssige Stoffmenge n s, ausgedrückt in Mol oder Gramm, die ein reales heterogenes System im Vergleich zum Referenzsystem, bezogen auf die Grenzflächenfläche bzw. auf die Oberfläche, aufweist des Adsorbens A. Es wird angenommen, dass das Vergleichssystem die gleichen intensiven Parameter (T, P, C i) und das gleiche Volumen (V = V α + V β) wie das reale System aufweist (Abb. 4). .
Г = (n - n α - n β)/A = n s /A 3.11
Überschüssige thermodynamische Funktionen des Übergangsbereichs eines realen Systems (wir bezeichnen sie mit dem Index s) können geschrieben werden als
U s = U – U α – U β , n s = n – n α – n β , S s = S – S α – S β usw.
Experimentelle Adsorptionsmessungen geben immer die Adsorption genau als Überschuss der Komponente im realen System im Vergleich zum gewählten Referenzsystem an. Um beispielsweise bei der Adsorption von Gas an einem festen Adsorptionsmittel oder bei der Adsorption von Komponenten an einer festen Phase Adsorptionswerte zu ermitteln, bestimmen Sie die Änderung der Anfangskonzentrationen des Adsorbats nach dem Kontakt der Phasen α und β
n i s = V(C i o - C i),
Wo C i o– Anfangskonzentration der i-ten Komponente, C ich– Konzentration der i-ten Komponente nach Herstellung des Gleichgewichts zwischen den Kontaktphasen. Es wird angenommen, dass die Lautstärke Vändert sich nicht. Allerdings ist die Konzentration ich Komponente C ich, experimentell erhalten, wird in Volumen bestimmt V'über der Phasengrenzfläche ohne Berücksichtigung des Volumens des inhomogenen Bereichs der Übergangsschicht V α an der Grenzfläche, wo die Konzentration ist C ich α. Aufgrund der Existenz einer ungleichmäßigen Region in einem realen System kann das Gesamtvolumen des Systems als dargestellt werden V = V’ + V α. Alle Menge ich-te Komponente C i o wird auf diese beiden Bände verteilt:
V C i o = V’ C i + V α C i α ,
und die Anzahl der Mol der Komponente ich, adsorbiert an der Grenzfläche, wird gleich sein
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Diese. Die experimentell ermittelte Adsorption ist der Überschuss der i-ten Komponente im Volumen V α im Vergleich zur Menge dieser Komponente im gleichen Volumen weit entfernt von der Phasengrenzfläche. Diese Art der Adsorption wird Gibbs-Adsorption genannt. .
V α C i α als vollständiger Inhalt bezeichnet ich- Komponente in der Adsorptionsschicht. Im Bereich sehr geringer Konzentrationen C ich im Volumen V'Änderung V α C i Gleichung (3.2) kann vernachlässigt und der Messwert berücksichtigt werden V α C i α Vollständiger Inhalt ich- Komponente in der Adsorptionsschicht, beispielsweise bei der Gasadsorption an einem festen Adsorbens bei niedrigen Drücken.
Thermodynamik von Adsorptionsprozessen.
| Parametername | Bedeutung |
| Thema des Artikels: | Thermodynamik von Adsorptionsprozessen. |
| Rubrik (thematische Kategorie) | Ausbildung |
Grundlegende Definitionen und Methoden zur Klassifizierung von Adsorptionsprozessen.
Adsorption bezieht sich auf Phänomene, die aufgrund einer spontanen Abnahme der Oberflächenenergie auftreten.
Adsorption– der Prozess der spontanen reversiblen oder irreversiblen Umverteilung der Komponenten eines heterogenen Systems zwischen der Oberflächenschicht und dem Volumen der homogenen Phase.
Bei Mehrkomponentensystemen wird vorzugsweise die Komponente, die die Grenzflächenspannung stärker reduziert, auf die Oberflächenschicht übertragen. Bei einkomponentigen Systemen kommt es bei der Bildung der Oberflächenschicht zu einer Änderung ihrer Struktur (eine bestimmte Ausrichtung von Atomen und Molekülen, Polarisation), genannt Autoadsorption.
Die dichtere Phase, in der Adsorptionswechselwirkungen lokalisiert sind, wird aufgerufen Adsorptionsmittel. Der zwischen dem Volumen der homogenen Phase und der Oberflächenschicht umverteilte Stoff wird mit dem Begriff „“ bezeichnet Adsorbatʼʼ.
In einigen Fällen ist der Adsorptionsprozess reversibel. In diesem Fall kann unter bestimmten Bedingungen ein Teil der adsorbierten Moleküle aufgrund molekularkinetischer Phänomene von der Oberflächenschicht in die Massenphase gelangen. Den umgekehrten Vorgang der Adsorption nennt man Desorption.
Methoden zur Klassifizierung von Adsorptionsprozessen.
Klassifizierung von Adsorptionsprozessen nach dem Aggregatzustand interagierender Phasen. Unter Berücksichtigung der Abhängigkeit vom Aggregatzustand benachbarter Phasen werden folgende Arten von Adsorptionsprozessen unterschieden:
Adsorption von Gasen an festen Adsorbentien;
Adsorption gelöster Stoffe an den Grenzflächen „fest-flüssig“ und „flüssig-flüssig“;
Adsorption von Tensiden an der Flüssigkeit-Gas-Grenzfläche.
Klassifizierung von Adsorptionsprozessen nach dem Mechanismus der Wechselwirkung zwischen Adsorbens und Adsorbat. Adsorption kann als Wechselwirkung von Adsorbatmolekülen mit den aktiven Zentren des Adsorbens betrachtet werden. Nach dem Mechanismus ihrer Wechselwirkung werden folgende Adsorptionsarten unterteilt:
1) physikalische (molekulare) Adsorption– Die Wechselwirkung zwischen den Molekülen des Adsorbats und des Adsorbens erfolgt aufgrund von Van-der-Waals-Kräften, Wasserstoffbrückenbindungen (ohne chemische Reaktionen);
2) chemische Adsorption (Chemisorption)– Die Anlagerung von Adsorbatmolekülen an die aktiven Zentren des Adsorbens erfolgt durch chemische Reaktionen verschiedener Art (mit Ausnahme von Ionenaustauschreaktionen);
3) Ionenaustauschadsorption (Ionenaustausch) – Umverteilung der Adsorbatsubstanz zwischen der Lösung und der festen Phase (Ionenaustauscher) gemäß dem Mechanismus der Ionenaustauschreaktionen.
Zur quantitativen Beschreibung von Adsorptionsprozessen werden zwei Größen verwendet.
1) Absolute Adsorption– Menge (Mol) oder Masse (kg) des Adsorbats pro Flächeneinheit oder Masse des Adsorbens. Bezeichnung – A; Dimension: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Gibbs-Adsorption (Überschuss).– Überschuss an Adsorbatsubstanz in einer Oberflächenschicht einer bestimmten Dicke im Vergleich zu ihrer Menge im Volumen der homogenen Phase, pro Flächeneinheit oder Masse des Adsorbens. Bezeichnung – G; Dimension: mol/m 2, mol/kᴦ.
Der Zusammenhang zwischen absoluter und überschüssiger Adsorption lässt sich anhand der Gleichung veranschaulichen:
Г = А – с * h (3.1)
wobei c die Gleichgewichtskonzentration des Stoffes im Volumen der Phase ist, mol/m3;
h ist die Dicke der Oberflächenschicht, die herkömmlicherweise mit 10 -9 m angenommen wird.
In heterogenen Mehrkomponentensystemen gilt bei Umverteilung der einen oder anderen Komponente zwischen dem Volumen der homogenen Phase und der Oberflächenschicht die Gleichung für die überschüssige innere Energie der Oberfläche:
U = T * S + s * s + Sm i * n i (3.2)
Wenn wir alle Terme der Gleichung auf die Flächeneinheit der Grenzflächenoberfläche reduzieren, erhalten wir:
U s = T * S s + s + Sm i * Г i (3.3)
wobei Г i = n i / s der Überschuss der i-ten Komponente in der Oberflächenschicht ist, d. h. Gibbs-Adsorption.
Für ein Einkomponentensystem hat Gleichung (3.3) die Form:
G s = s + m * G (3.4)
wobei G s = U s - T * S s – Gibbs-Energie der Oberfläche oder die Arbeit zur Erzeugung einer Flächeneinheit;
m * G – Verdichtung der adsorbierten Substanz in der Oberflächenschicht.
Basierend auf Gleichung (3.4) können wir schließen, dass während der Adsorption die Arbeit zur Schaffung einer Grenzflächenoberfläche aus der Arbeit der Oberflächenbildung (Aufbrechen kohäsiver Bindungen im Volumen der Adsorbatphase) und der Verdichtung der Substanz in der Oberflächenschicht besteht.
Im Zustand des dynamischen Gleichgewichts zwischen Adsorbens und Adsorbat, der Änderung der Gibbs-Energie des heterogenen Systems ΔG = 0, wird die Thermodynamik des Adsorptionsprozesses durch die genannte Gleichung beschrieben Grundlegende Adsorptionsgleichung von Gibbs:
Ds = SГ i * dm i (3.5)
Diese Gleichung ist universell, da sie für alle Arten von Adsorptionsprozessen gilt
Sonderfälle der Gibbs-Adsorptionsgleichung.
1) Adsorption aus Lösungen.
Für das chemische Potenzial der i-ten Komponente des Systems während der Adsorption an den Grenzflächen „Flüssigkeit – fester Adsorber“ und „Flüssigkeit – Gas“ gelten folgende Gleichungen:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
wobei m i 0 das chemische Potential der i-ten Komponente des Systems unter Standardbedingungen ist;
a i ist die Aktivität der i-ten Komponente des Systems unter Standardbedingungen.
Auf dieser Grundlage hat die Gibbs-Adsorptionsgleichung die Form:
Г i = - a i / R*T * (ds / da i) (3.8)
Für Lösungen von Nichtelektrolyten nehmen wir a i = c i, dann:
Г i = - с / R*T * (ds / dс) (3.9)
Für Elektrolytlösungen:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
wobei с ± die durchschnittliche Ionenkonzentration der Lösung ist;
n ist der stöchiometrische Koeffizient.
2) Adsorption von Stoffen aus der Gasphase.
Gemäß der Mendeleev-Clayperon-Gleichung:
Р = с * R*T (3.11)
In diesem Zusammenhang lautet die Gibbs-Gleichung für die Adsorption von Gasen an festen Adsorbentien wie folgt:
Г i = - Р / R*T * (ds / dР) (3.12)
In der Praxis ermöglicht die Gibbs-Adsorptionsgleichung, basierend auf Oberflächenspannungsmessungen bei verschiedenen Werten der Flüssigkeitskonzentration oder des Gleichgewichtsgasdrucks, die Adsorptionsmenge von Substanzen in der Grenzflächenschicht zu berechnen, für die die Oberflächenspannung bestimmt wird.
Thermodynamik von Adsorptionsprozessen. - Konzept und Typen. Einordnung und Merkmale der Kategorie „Thermodynamik von Adsorptionsprozessen“. 2017, 2018.
Adsorption als spontane Konzentration von Molekülen auf einer Oberfläche geht mit einer Abnahme der Entropie des Systems einher. Denn das Kriterium für die Spontaneität des Prozesses ist
∆H - T · ∆S = ∆G< 0,
dann ist Adsorption nur bei ∆H möglich< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Mit steigender Temperatur verschiebt sich das Gleichgewicht in Richtung des endothermen Prozesses, also der Desorption.
Adsorption auf einer festen Oberfläche
1. Monomolekulare Adsorption.
Nach Langmuirs Theorie interagieren Adsorbensmoleküle mit der Oberfläche des Adsorbens und bilden letztendlich eine monomolekulare Schicht. Dabei handelt es sich um den Füllungsgrad () der Oberfläche mit dem adsorbierten Stoff bei der Adsorption aus der Gasphase
aus Flüssigkeit
wobei K die Gleichgewichtskonstante (Adsorptionskonstante) ist;
p ist der Partialdruck des adsorbierten Gases;
c ist die Konzentration der adsorbierten Substanz.
Die Abhängigkeit von β von p (oder c) wird durch den Graphen (Adsorptionsisotherme, T = const) in Abb. dargestellt. 1.3.

Reis. 1.3. Grad der Oberflächenfüllung mit adsorbierter Substanz
Bei niedrigen Konzentrationen und Partialdrücken ist die Adsorption proportional zur Konzentration bzw. zum Partialdruck:
R<< 1, β ≈ К· r ilis<< 1, β ≈ К· s, d.h. der Anfangsabschnitt der Isotherme ist annähernd linear und tan α = K (tg α wird durch die Steigung der Kurve bei p (oder c) → 0: oder bestimmt).
If ist die Anzahl der Mol adsorbierter Substanz pro 1 g Adsorbens; - die maximal mögliche Molzahl der adsorbierten Substanz pro 1 g Adsorbens („Monoschichtkapazität“)
Einsetzen von β in Gleichung (1.3) (für den Fall der Adsorption aus der Gasphase die Konzentration). Mit in Gleichungen sollte durch Druck ersetzt werden R), wir bekommen:
 (1.6)
(1.6)
Da und K in einem gegebenen Adsorbens-Adsorbens-Paar Konstanten sind (bei T=const), dann kann man durch Abhängigkeit finden ZU(Abb. 1.4).

Reis. 1.4. Grafische Lösung der Adsorptionsgleichung
erhalten durch Extrapolation der experimentellen linearen Abhängigkeit auf () = 0; und seitdem , .
Mit dem Wert lässt sich die spezifische Oberfläche des Adsorbens bestimmen UD (in m 2 pro 1 g Adsorbens), wenn die von einem Molekül des Adsorbens auf der Oberfläche eingenommene Fläche ω bekannt ist (bestimmt aus der Größe des Moleküls):
UD = · ω · Na, (1.7)
wobei Na die Avogadro-Zahl ist (Na = 6,02 · 10 · 23).
Der bekannte Wert von UD kann wiederum zur Berechnung von ω jeder Substanz basierend auf ihrer Adsorption an einem bestimmten Adsorptionsmittel verwendet werden.
2. Polymolekulare Adsorption.
Gleichung (1.5) beschreibt eine Kurve mit Sättigung, d.h. bei
p (oder c) → ∞ tendiert zum Grenzwert gleich (Abb. 1.5,a).

Abb.1.5. Adsorptionsisothermen:
a – Adsorption mit Sättigung; b – polymolekulare Adsorption
In einigen Fällen sehen die Adsorptionsisothermen jedoch wie in Abb. 1,5, b, d.h. erreicht selbst bei hohem p (oder c) nicht den Grenzwert.
Abhängigkeiten der in Abb. gezeigten Art. 1.5,b entsprechen der polymolekularen Adsorption. Solche Isothermen sind in der Regel charakteristisch für Stoffe mit starken intermolekularen Wechselwirkungen (z. B. Wasser). Wenn die Adsorptionszentren auf der Oberfläche des Adsorbens besetzt sind (die monomolekulare Schicht ist gesättigt), erfolgt die „Landung“ der nächsten Adsorbatmoleküle aufgrund intermolekularer Wechselwirkungen mit bereits adsorbierten Molekülen (Abb. 1.6). Die Wärme einer solchen Adsorption liegt im absoluten Wert nahe beieinander, hat jedoch ein entgegengesetztes Vorzeichen wie die Verdampfungswärme der entsprechenden Flüssigkeit (denken Sie darüber nach, warum).

Abb.1.6. Adsorptionsschema:
a - monomolekulare Adsorption; b – polymolekulare Adsorption
Je näher wir kommen R Bei Erreichen des Sättigungsdampfdrucks des adsorbierten Stoffes beginnt dieser an der Oberfläche des Adsorbens zu kondensieren, wodurch er mit zunehmendem Druck schnell zunimmt R.
Im Falle einer Wechselwirkung zwischen zwei Atomen:
U – Wechselwirkungsenergie;
U = U VOR. + U RÜCKKEHR
 - Lennard-Jones-Gleichung
, c, b, m = const
- Lennard-Jones-Gleichung
, c, b, m = const
Bei der Wechselwirkung von Atomen mit einer festen Oberfläche ist es notwendig, alle Wechselwirkungen zusammenzufassen.
x – Abstand zur Oberfläche
r – Wirkungsradius der Anziehungskräfte
dV – Lautstärke
n – Anzahl der Oberflächenmoleküle
U ANZEIGEN. – Energie der Adsorptionswechselwirkung



Bei der Adsorption erhöht sich die Anziehungskraft. Und bei unpolar-unpolarer Wechselwirkung ist die Adsorption überwiegend in Vertiefungen lokalisiert.
Elektrostatische Interaktion.
Polares Adsorbens – unpolares Adsorbat
Unpolares Adsorbens – polares Adsorbat
Polares Adsorbens – polares Adsorbat.
M  Das Adsorbatmolekül wird als Dipol dargestellt, und das Adsorbens wird als Leiter dargestellt, in dem das Adsorbatmolekül einen Dipolspiegel symmetrisch zum gegebenen Spiegel induziert.
Das Adsorbatmolekül wird als Dipol dargestellt, und das Adsorbens wird als Leiter dargestellt, in dem das Adsorbatmolekül einen Dipolspiegel symmetrisch zum gegebenen Spiegel induziert.
X – Abstand zur Mitte
Im Zusammenspiel entstehen Potenziale:
 ,
,
 - Dipolmoment.
- Dipolmoment.
Das Potenzial tendiert dazu, den Maximalwert anzunehmen, d.h. Dipole neigen dazu, sich senkrecht zur Oberfläche auszurichten.
Da eine Temperaturerhöhung das Wachstum der Brownschen Bewegung fördert, führt sie zu einer Hemmung des Adsorptionsprozesses.
Bei elektrostatischer Wechselwirkung ist das Adsorbat überwiegend an den Vorsprüngen lokalisiert.
Grundlegende Adsorptionsgleichung.
Bei der Adsorption kommt es zu einer Umverteilung der Komponente, das heißt, das chemische Potenzial verändert sich. Der Adsorptionsprozess kann als Umwandlung von Oberflächenenergie in chemische Energie betrachtet werden.
Schichtvolumen = 0, dann gilt die verallgemeinerte Gleichung der Hauptsätze I und II der Thermodynamik:
T = const; (1) = (2) => 

Für ein Zweikomponentensystem:
,
 ,
,
 =>
=>
=>
 - Gibbs-Adsorptionsgleichung
.
- Gibbs-Adsorptionsgleichung
.
Für den Fall der TV-Adsorption. Körper - Gas: , 
 ,
,

 - Isotherme
- Isotherme
 - Isobare
- Isobare
 - isopyknal
- isopyknal
 - isostere
- isostere
Isotherme, Isopykne, Isostere hängen miteinander zusammen.
Weil Adsorptionsfunktion 



Henry-Isotherme Langmuir-Isotherme



Thermodynamik. Adsorption.
Für kondensierte Materie:
 ,
,
 ,
,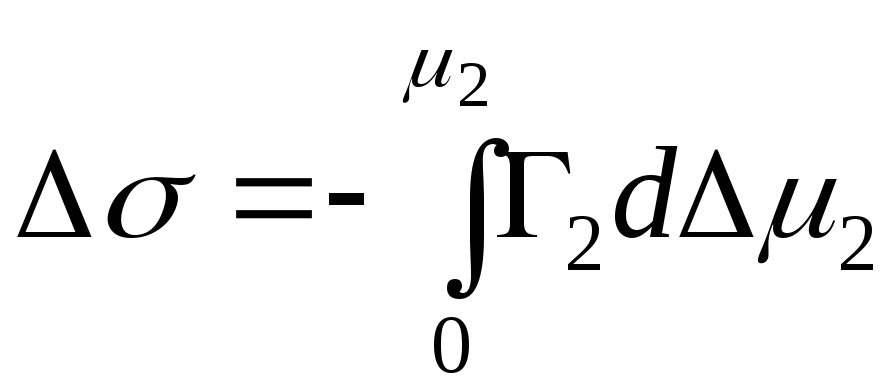
 - integrale Änderung der Gibbs-Energie
.
- integrale Änderung der Gibbs-Energie
.

P – Druck über einer gekrümmten Fläche, Р S – Druck über einer ebenen Fläche
 - Adsorptionspotential
- Adsorptionspotential
Differenzielle Änderung der Entrapie 
 , Г = const
, Г = const
- differenzielle Entropieänderung
- Differenzielle Adsorptionsenthalpie
 - isosterische Adsorptionswärme
- isosterische Adsorptionswärme
 - Kondensationswärme
- Kondensationswärme
 - Nettoadsorptionswärme
- Nettoadsorptionswärme

 ,
,




Qa – integrale Adsorptionswärme, 
Qra – integrale Nettoadsorptionswärme,

Henrys Gleichung
Die Untersuchung der Adsorption wird durch die Heterogenität der Oberfläche erschwert, sodass die einfachsten Gesetze für homogene Oberflächen gelten.
Betrachten wir die Wechselwirkung von Gasen mit einer festen Oberfläche, wenn ein Gas von einem Gleichgewichtszustand im Volumen in einen Gleichgewichtszustand auf der Oberfläche übergeht. Dieser Fall ist analog zum Gleichgewicht von Gasen in einem Schwerefeld.
 ,
,
 ,
=>
,
=> -Henrys Gleichung
-Henrys Gleichung
 - Verteilungskoeffizient
- Verteilungskoeffizient
Während des Adsorptionsprozesses kommt es zu einer Änderung der chemischen Potentiale.
Für die Bulk-Phase: 
Für Gas an der Oberfläche: 
Im Gleichgewicht  , d.h.
, d.h.

In Henrys Gleichung hängt die Konstante nicht von der Konzentration ab
Die Henry-Gleichung gilt im Bereich niedriger Drücke und Konzentrationen. Mit zunehmender Konzentration sind zwei Arten von Abweichungen vom Henry’schen Gesetz möglich:
1 – positive Abweichungen, D nimmt ab, A nimmt ab
2 – negative Abweichungen, D – Zuwächse, A – Zuwächse.
Die Art der Abweichung wird durch das Vorherrschen der einen oder anderen Art der Adsorbens-Adsorbat-Wechselwirkung bestimmt.
Bei starker adhäsiver Wechselwirkung steigen die Aktivitätskoeffizienten – eine positive Abweichung. Bei kohäsiven Wechselwirkungen werden negative Abweichungen beobachtet.
Monomolekulare Adsorption.
Langmuir-Isotherme.
Die einfachsten Muster wurden in Henrys Theorie erhalten. Langmuir schlug eine Theorie vor, nach der Adsorption als quasi-chemische Reaktion betrachtet wird. Dabei:
Die Oberfläche ist energetisch homogen.
Die Adsorption erfolgt lokal, jedes Adsorptionszentrum interagiert mit einem Adsorbatmolekül.
Adsorbatmoleküle interagieren nicht miteinander.
Monoschichtadsorption.

 - Oberfläche,
- Oberfläche,  - Adsorbat,
- Adsorbat,  - Adsorptionskomplex.
- Adsorptionskomplex.
 , dann die Konzentration der Adsorptionsstellen:
, dann die Konzentration der Adsorptionsstellen:  ,
, - Begrenzung der Adsorption.
- Begrenzung der Adsorption.
 , dann ist die Reaktionskonstante:
, dann ist die Reaktionskonstante: 

 - Langmuir-Gleichung.
- Langmuir-Gleichung.
Abhängigkeit der Adsorption von der Konzentration
1 )
)
 ,
,
2) Bereich hoher Konzentrationen
 - Begrenzung der Adsorption, Bildung einer monomolekularen Schicht
- Begrenzung der Adsorption, Bildung einer monomolekularen Schicht
Für Gibbs-Energie: .
g ist der Entropiefaktor.
Im Fall der Henry-Isotherme charakterisiert die Gibbs-Energie den Übergang des Adsorbats vom Standardzustand in der Masse in den Standardzustand an der Oberfläche. Im Fall der Langmuir-Isotherme  charakterisiert den Grad der Affinität zwischen dem Adsorbens und dem Adsorbat.
charakterisiert den Grad der Affinität zwischen dem Adsorbens und dem Adsorbat.
 gefunden aus der Van't-Hoff-Isobare.
gefunden aus der Van't-Hoff-Isobare.

 , Dann
, Dann  , von hier
, von hier  .
.

 - Grad der Oberflächenfüllung.
- Grad der Oberflächenfüllung.




 - Anzahl der freien Plätze,
- Anzahl der freien Plätze,  - Anzahl der belegten Plätze.
- Anzahl der belegten Plätze.
 ,
,

Diese. Im Bereich hoher Konzentrationen ist die Zahl der freien Stellen umgekehrt proportional zur Adsorbatmenge.
Adsorption eines Gasgemisches auf einer homogenen Oberfläche.
In diesem Fall wird der Adsorptionsprozess als zwei parallele Reaktionen betrachtet.
 (1)
(1)

 (2)
(2)


Adsorption eines Gasgemisches auf einer ungleichmäßigen Oberfläche.
Bei einer ungleichmäßigen Oberfläche kann man sich nicht auf durchschnittliche Füllungen beschränken.
Durch die Konkurrenz ist die Lokalisierung unterschiedlicher Adsorbate in Gebieten unterschiedlichen Typs möglich.
In diesem Fall die Relation  .
.
 ,
,
 - Sättigungsdampfdruck des Adsorbats.
- Sättigungsdampfdruck des Adsorbats.
 ,
,
 - Adsorptionswärme.
- Adsorptionswärme.
„+“ – Symbate-Abhängigkeit, „-“ – Antibate-Abhängigkeit, „N“ – keine Korrelation.
„+“ – die Adsorption erfolgt nach dem gleichen Mechanismus. In den energetisch günstigsten Bereichen wird überwiegend Gas mit hoher Affinität zur Oberfläche adsorbiert.
„-“ – Adsorption erfolgt über verschiedene Mechanismen und bis zu einem bestimmten Zeitpunkt gibt es keine Konkurrenz um die Oberfläche.
Die monomolekulare Adsorption erfolgt überwiegend bei der physikalischen Adsorption von Gasen bei niedrigen Werten P sowie an der Grenzfläche Flüssigkeit/Gas.
Polymolekulare Adsorption.
BET-Theorie(Brunauer, Emmett, Teller).
Reicht die Bildung einer Monoschicht nicht aus, um die Oberflächenenergie zu kompensieren, ist die Adsorption polymolekular und kann als Ergebnis einer erzwungenen Kondensation unter Einwirkung von Oberflächenkräften betrachtet werden.
Kernpunkte:
Wenn ein Adsorbatmolekül auf eine besetzte Stelle trifft, wird ein Mehrfachsatz gebildet.
Je näher wir kommen P Zu P S die Zahl der freien Adsorptionsstellen nimmt ab. Zunächst nimmt die Zahl der Plätze, die mit Einzel-, Doppel-, etc. belegt werden, zu und dann ab. in Sätzen.
Bei P =P S Adsorption geht in Kondensation über.
Es gibt keine horizontalen Wechselwirkungen.
Für die erste Schicht ist die Langmuir-Isotherme erfüllt.
Die Oberfläche wird als eine Reihe von Adsorptionsstellen betrachtet. Es gilt die Bedingung des dynamischen Gleichgewichts: Die Kondensationsrate an freien Orten ist gleich der Verdunstungsrate an besetzten Orten.
a ist der Kondensationskoeffizient (der Anteil der auf der Oberfläche kondensierten Moleküle);
 ,
,
Zm – maximale Anzahl freier Plätze.
 - Frequenz der Atomschwingungen in Richtung senkrecht zur Oberfläche.
- Frequenz der Atomschwingungen in Richtung senkrecht zur Oberfläche.
Für die erste Schicht gelten dynamische Gleichgewichtsbedingungen:



 , Dann
, Dann
 - Langmuir-Gleichung.
- Langmuir-Gleichung.
Für die zweite Schicht gilt: 
Für die i-te Schicht: 
Der Einfachheit halber wird angenommen, dass a und ν für alle Schichten außer der ersten gleich sind. Für alle Schichten außer der ersten ist die Adsorptionswärme konstant. Für die letzte Schicht ist die Adsorptionswärme gleich der Kondensationswärme. Als Ergebnis wurde die Gleichung erhalten
 (*)
(*)
C– konstant,
Im Fall der BET-Theorie die Konstante MIT charakterisiert die Gibbs-Energie der reinen Adsorption. Die Gleichung enthält nur eine Konstante und diese Gleichung ist auch für die Bestimmung der spezifischen Oberfläche des Adsorbens sehr wichtig.
Da bei der Adsorption Wärme freigesetzt wird, werden spezifische Oberflächen bei niedrigen Temperaturen bestimmt.
 ????????????
????????????


Der Hauptnachteil der Theorie– Vernachlässigung horizontaler Wechselwirkungen zugunsten vertikaler.
Die Gleichung gilt im Bereich  von 0,05 bis 0,3.
von 0,05 bis 0,3.
Wo  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – die Adsorbat-Adsorbat-Wechselwirkung wird beeinflusst.
> 0,3 – die Adsorbat-Adsorbat-Wechselwirkung wird beeinflusst.
Berücksichtigung von Adsorbat-Adsorbat-Wechselwirkungen.
Wechselwirkungen treten auf, wenn verzweigte Moleküle oder Moleküle an einer unpolaren Oberfläche adsorbiert werden. Kann Mitarbeiter bilden. In diesem Fall ändert sich die Form der Adsorptionsisothermen.
A  das Adsorptionsmittel ist nicht polar.
das Adsorptionsmittel ist nicht polar.
Diagramm 1 entspricht schwachen Adsorbat-Adsorbat-Wechselwirkungen und starken Adsorbat-Adsorbens-Wechselwirkungen.
Diagramm 2 entspricht starken Adsorbat-Adsorbat- und starken Adsorbat-Adsorbens-Wechselwirkungen.
Diagramm 3 entspricht einer starken Adsorbat-Adsorbat-Wechselwirkung und einer schwachen Adsorbat-Adsorbens-Wechselwirkung.
 ,
,

Bei Wechselwirkungen zwischen Adsorbatmolekülen müssen Änderungen der Aktivitätskoeffizienten berücksichtigt werden. Und diese Gleichung wird geschrieben als:
 - Frunkin-, Fowler-, Guggenheim-Gleichung.
- Frunkin-, Fowler-, Guggenheim-Gleichung.
k– Anziehungskonstante.
Polyanys Potentialtheorie.
Diese Theorie leitet keine Art von Adsorptionsisotherme ab, ermöglicht jedoch die Berechnung von Isothermen bei einer anderen Temperatur.
Adsorption- Dies ist das Ergebnis der Anziehung des Adsorbats an die Oberfläche des Adsorbens aufgrund der Wirkung des Adsorptionspotentials, das nicht von der Anwesenheit anderer Moleküle abhängt und vom Abstand zwischen der Oberfläche und dem Adsorbatmolekül abhängt.
 ,
,
 - Adsorptionspotential.
- Adsorptionspotential.
Da die Oberfläche ungleichmäßig ist, wird der Abstand durch das Adsorptionsvolumen ersetzt  .Adsorptionsvolumen ist das zwischen der Oberfläche und dem Punkt eingeschlossene Volumen, das einem bestimmten Wert entspricht
.Adsorptionsvolumen ist das zwischen der Oberfläche und dem Punkt eingeschlossene Volumen, das einem bestimmten Wert entspricht  .
.
Adsorptionspotential ist die Arbeit für die Übertragung von 1 Mol Adsorbat außerhalb eines bestimmten Adsorptionsvolumens an einen bestimmten Punkt des Adsorptionsvolumens (oder die Arbeit für die Übertragung von 1 Mol gesättigtem Dampf eines Adsorbats, das sich in Abwesenheit eines Adsorptionsmittels im Gleichgewicht mit einem flüssigen Adsorbat befindet). in eine Dampfphase im Gleichgewicht mit dem Adsorbens übergeht).

Charakteristische Kurve
 - Adsorptionspotential,
- Adsorptionspotential, 
Für ein gegebenes Adsorbens und verschiedene Adsorbate gilt Folgendes: 
Für verschiedene Arten von Adsorbaten  ,
,
Wo  Potentiale für Adsorptionsisothermen bei relativen Drücken
Potentiale für Adsorptionsisothermen bei relativen Drücken  für Adsorbat 1 und für Adsorbat 2. Dieses Verhältnis ist ein konstanter Wert.
für Adsorbat 1 und für Adsorbat 2. Dieses Verhältnis ist ein konstanter Wert.
 - Affinitätskoeffizient
- Affinitätskoeffizient
Theorie der Kapillarkondensation.
Der Ablauf des Adsorptionsprozesses hängt maßgeblich von der Struktur des porösen Körpers ab.
|
Mikroporös | |
|
Übergangsweise porös | |
|
Makroporös |
Bei mikroporösen Sorptionsmitteln überlappen sich die Felder der Adsorptionskräfte. Bei makroporösen Sorptionsmitteln fungieren Poren als Transportkanäle. Kondensationsprozesse sind in übergangsweise porösen Körpern am bedeutendsten. Ab bestimmten Werten beginnt die Kapillarkondensation P Und  , wenn ein Teil der Oberflächenenergie bereits kompensiert wurde. Voraussetzung ist, dass die Oberfläche selbstbenetzend ist. Der Vorgang wird beschrieben Thompson-Kelvin-Gleichung.
, wenn ein Teil der Oberflächenenergie bereits kompensiert wurde. Voraussetzung ist, dass die Oberfläche selbstbenetzend ist. Der Vorgang wird beschrieben Thompson-Kelvin-Gleichung.

 - Bei Benetzung liegt der Krümmungsmittelpunkt in der Gasphase.
- Bei Benetzung liegt der Krümmungsmittelpunkt in der Gasphase.
Bei der Kapillarkondensation hat die Adsorptionsisotherme eine hysteretische Form. Der untere Zweig entspricht dem Adsorptionsprozess und der obere Zweig entspricht dem Desorptionsprozess.
Alle Arten von Poren können auf drei Arten reduziert werden:
|
Konisch |
Zylindrisch mit einem geschlossenen Ende |
Zylindrisch mit zwei offenen Enden |
|
Die Prozessbefüllung erfolgt vom Porenboden aus. Die Adsorptionsisotherme und die Desorptionsisotherme fallen in diesem Fall zusammen, da der Adsorptionsprozess von einer Kugel ausgeht und der Desorptionsprozess ebenfalls mit dem Verschwinden einiger Kugeln beginnt.
↓ |
Es gibt keine Hysterese. Vorwärts- und Rückwärtshub werden durch die Gleichung beschrieben:
|
Es gibt nirgendwo einen Boden, die Füllung der Pore verläuft entlang der Wände des Zylinders.
Zylinder: Die Isotherme hat ein hystereseartiges Aussehen.
↓ |


 - Kugel,
- Kugel, ,
,



IN  Unter Benetzungsbedingungen findet die Kondensation bei niedrigeren Drücken statt, was energetisch günstig ist. Aus dem Desorptionszweig werden Porengrößenverteilungskurven erhalten.
Unter Benetzungsbedingungen findet die Kondensation bei niedrigeren Drücken statt, was energetisch günstig ist. Aus dem Desorptionszweig werden Porengrößenverteilungskurven erhalten.
Das Maximum der Differentialkurve ist gegenüber dem Wendepunkt der Integralkurve nach links verschoben. Das Gesamtvolumen kleiner Poren ist klein, weist aber große Oberflächen auf. Mit zunehmender Porengröße nimmt ihr Volumen zu  , und die Gegend ist wie
, und die Gegend ist wie  Dadurch wird eine Verschiebung des Maximums der Differenzkurve beobachtet.
Dadurch wird eine Verschiebung des Maximums der Differenzkurve beobachtet.
Adsorption an der Fest-Flüssigkeits-Grenzfläche.
Bei der Adsorption an der Fest-Gas-Grenzfläche haben wir eine Komponente vernachlässigt. Bei der Adsorption an der Fest-Flüssigkeits-Grenzfläche verdrängt das Adsorbat Lösungsmittelmoleküle von der Oberfläche des Adsorbens.
 ,
,
Die Gleichung ist richtig:

 ,
,
N 1, N 2 – Stoffmengenanteile von Lösungsmittel und Komponente, N 1 + N 2 = 1, dann
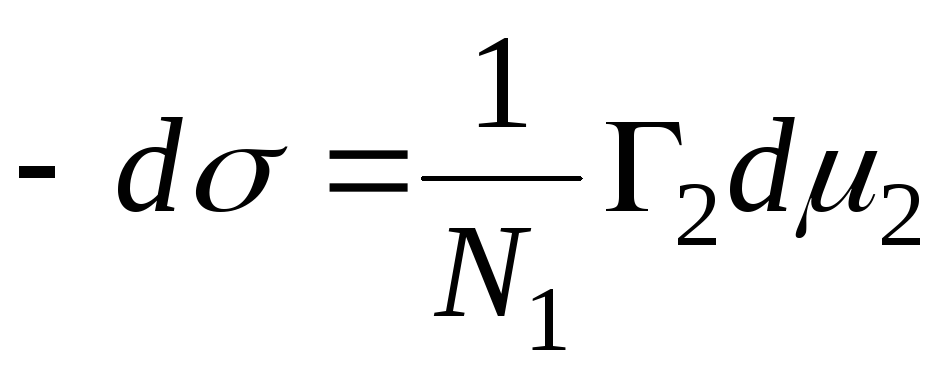 ,
=>
,
=>
 , dann ist die Adsorptionsgleichung für die Fest-Flüssigkeits-Grenzfläche.
, dann ist die Adsorptionsgleichung für die Fest-Flüssigkeits-Grenzfläche.
Adsorption (G) > 0 at  <
0
<
0
Wenn die Werte  denn die Komponente und das Lösungsmittel sind sehr unterschiedlich, in diesem Fall die Abhängigkeit G aus N hat ein Extremum bei dem Wert N
~ 0,5.
denn die Komponente und das Lösungsmittel sind sehr unterschiedlich, in diesem Fall die Abhängigkeit G aus N hat ein Extremum bei dem Wert N
~ 0,5.
E  Wenn
Wenn  nahe beieinander liegen, in diesem Fall kann sich das Vorzeichen der Adsorption ändern. Sucht G aus N kreuzt die x-Achse
nahe beieinander liegen, in diesem Fall kann sich das Vorzeichen der Adsorption ändern. Sucht G aus N kreuzt die x-Achse
Schnittpunkt der Funktion G(N) mit der x-Achse heißt Adsorptionsazeotrop. Dies bedeutet, dass die beiden Komponenten an einem bestimmten Adsorbens nicht getrennt werden können.
Gleichung der Adsorptionsisotherme mit Austauschkonstante.
Bei der Adsorption an der Fest-Flüssigkeits-Grenzfläche kommt es zu einer ständigen Umverteilung der Komponenten zwischen der Oberfläche des Adsorbens und dem Volumen der Lösung.
 - Komponenten (- - beziehen sich auf die Oberfläche)
- Komponenten (- - beziehen sich auf die Oberfläche)

 ,
,
 ,
, .
.
 ,
,


Adsorption an der Flüssigkeits-Gas-Grenzfläche
R  Betrachten wir die Änderung des Konzentrationsprofils beim Überqueren der Flüssigkeits-Gas-Grenzfläche. Lassen Sie Komponente 2 flüchtig sein.
Betrachten wir die Änderung des Konzentrationsprofils beim Überqueren der Flüssigkeits-Gas-Grenzfläche. Lassen Sie Komponente 2 flüchtig sein.
Cs – Konzentration in der Oberflächenschicht.
Basierend auf der Definition der überschüssigen Adsorption

Wenn die Komponente nicht flüchtig ist, wird der Adsorptionswert wie folgt geschrieben:
P  ri
ri 

In Gl.  Die Natur eines Stoffes wird durch sein Derivat beschrieben
Die Natur eines Stoffes wird durch sein Derivat beschrieben  .
.
Die Oberflächenspannungsisotherme kann die Form 1 oder 2 haben:
1 – Tenside
2 – Tenside
Unter Oberflächenaktivität g versteht man die Fähigkeit von Stoffen, die Oberflächenspannung in einem System zu verringern.

 - Dicke der Oberflächenschicht
- Dicke der Oberflächenschicht
C S– Konzentration der Komponente in der Oberflächenschicht
MIT– Volumenkonzentration
Für eine homologe Reihe gibt es eine Regel:
 - Traubo-Duclos-Regel
- Traubo-Duclos-Regel
Für eine homologe Reihe sieht die Adsorptionsisotherme folgendermaßen aus:
Anstelle von A schreiben wir G, da die Adsorption in der Oberflächenschicht zu stark ist.
Oberflächenspannungsisotherme:

 - Oberflächenspannung eines reinen Lösungsmittels.
- Oberflächenspannung eines reinen Lösungsmittels.
 - grundlegende Adsorptionsgleichung;
- grundlegende Adsorptionsgleichung;
 - Langmuir-Gleichung.
- Langmuir-Gleichung.
Lassen Sie uns sie gemeinsam lösen:

- Shishkovsky-Gleichung.
B– Konstante für die homologe Reihe.
A- Beim Übergang von einem Homologen zu einem anderen erhöht sich der Wert um das 3- bis 3,5-fache 
![]()
1 – Bereich mit geringen Konzentrationen
![]()
2 – durchschnittliche Konzentration
3 – monomolekulare Schicht
Tenside sind diphile Moleküle, d. h. umfassen eine polare Gruppe und einen unpolaren Kohlenwasserstoffrest.
o ist der polare Teil des Moleküls.
| - unpolarer Teil des Moleküls.
In einem polaren Lösungsmittel sind Tensidmoleküle so ausgerichtet, dass der polare Teil des Moleküls dem Lösungsmittel zugewandt ist und der unpolare Teil in die Gasphase gedrückt wird.
In Shishkovskys Gleichung  , sie ist für die homologische Reihe konstant.
, sie ist für die homologische Reihe konstant.
Der Tensideffekt beginnt mit zu erscheinen N>5. Bei Konzentrationen, die höher sind als die Konzentration der monomolekularen Schicht, kommt es in Tensidlösungen zur Mizellenbildung.
Mizelle– nennt man ein Aggregat amphiphiler Tensidmoleküle, deren Kohlenwasserstoffreste einen Kern bilden und deren polare Gruppen in die wässrige Phase überführt werden.
Mizellenmasse – Mizellenmasse.
H  Anzahl der Moleküle – Aggregationszahl.
Anzahl der Moleküle – Aggregationszahl.
Sphärische Mizellen
Bei der Mizellisierung stellt sich in der Lösung ein Gleichgewicht ein
CMC – kritische Konzentration der Mizellenbildung.
Da wir die Mizelle als separate Phase betrachten:
Für eine homologische Reihe gibt es eine empirische Gleichung:
A– Auflösungsenergie der funktionellen Gruppe.
B – Erhöhung des Adsorptionspotentials, Adsorptionsarbeit pro Methyleneinheit.
– Erhöhung des Adsorptionspotentials, Adsorptionsarbeit pro Methyleneinheit.
Das Vorhandensein eines Kohlenwasserstoffkerns in Mizellen bietet die Möglichkeit, dass in Wasser unlösliche Verbindungen in wässrigen Lösungen von Tensiden gelöst werden. Dieses Phänomen wird als Solubilisierung bezeichnet (was sich auflöst, ist das Solubilisat, das Tensid ist der Lösungsvermittler).
Der Schlamm kann völlig unpolar sein, sowohl polare als auch unpolare Teile enthalten und wie ein Tensidmolekül ausgerichtet sein.
In jedem Fall kommt es während der Solubilisierung zu einer Zunahme der Mizellenmasse und der Aggregationszahl, nicht nur aufgrund des Einschlusses von Solubilisat, sondern auch aufgrund einer Zunahme der Anzahl an Tensidmolekülen, die zur Aufrechterhaltung eines Gleichgewichtszustands erforderlich sind.
Die Solubilisierung ist umso effektiver, je niedriger das Molekulargewicht des Solubilisats ist.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
Die Wirksamkeit von Tensiden hängt vom CMC-Wert ab.
2D-Oberflächenschichtdruck

→ -Oberflächenspannungskräfte.
- zweidimensionaler Druck.
Die Oberflächenschicht ist eine Kraft, die dem Unterschied in der Oberflächenspannung einer Tensidlösung und einem reinen Lösungsmittel entspricht und auf eine saubere Oberfläche gerichtet ist.
Es stellt sich ein Gleichgewicht zwischen der Lösung und der Oberflächenschicht ein



Bei  Es gibt einen Bereich, in dem
Es gibt einen Bereich, in dem  hängt linear von der Konzentration ab.
hängt linear von der Konzentration ab.
G [mol/m2].
 -Fläche, die von einem Mol einer Substanz eingenommen wird
-Fläche, die von einem Mol einer Substanz eingenommen wird
Dann hat die zweidimensionale Druckisotherme die Form 
 - zweidimensionale Druckisotherme.
- zweidimensionale Druckisotherme.
Sucht  von S M:
von S M:

Bei  - Der zweidimensionale Druck steigt stark an. Bei
- Der zweidimensionale Druck steigt stark an. Bei  zweidimensional wird deformiert, was zu plötzlichem Wachstum führt
zweidimensional wird deformiert, was zu plötzlichem Wachstum führt  .
.
Ein Film, der auf beiden Seiten von identischen Phasen begrenzt wird, wird als doppelseitig bezeichnet. Bei solchen Filmen ist eine ständige Bewegung der Mutterlauge zu beobachten.
Filme mit einer Dicke von weniger als 5 nm werden als schwarze Filme bezeichnet.

Adsorptionsschichten müssen zwei Eigenschaften aufweisen: Viskosität und leichte Beweglichkeit, Fließfähigkeit und Elastizität.
Der Marangoni-Effekt ist selbstheilend.
Gibbs-Dreieck,  - Überdruck.
- Überdruck.
Der Film hat sich gedehnt und durch den Austritt eines Teils der Flüssigkeit dringen die Tenside in den freien Raum ein. Gibbs-Dreieck.
Die Wirkung der Adsorptionsstärke von Körpern.
Auf der Oberfläche der Folie befindet sich immer eine Adsorptionsschicht, für die dann
Langmuir-Gleichung:


 in zweidimensionalen Druck umwandeln
in zweidimensionalen Druck umwandeln
 - ein Analogon der Shishkovsky-Gleichung
- ein Analogon der Shishkovsky-Gleichung
Elektrokinetische Phänomene. Elektrische Doppelschicht (EDL).
Gelemholtz-Modell. Gouy-Chapman-Theorie.
1808 Flug

U – geformtes Rohr, tauchen Sie 2 Elektroden hinein. Das Gesetz der kommunizierenden Gefäße wird verletzt und es kommt zu einer Änderung des Flüssigkeitsspiegels im Rohr – elektrokinetische Phänomene.
Kinetische Phänomene:
Elektrophorese
Elektroosmose
Fluss-(Fluss-)Potenzial
Sedimentationspotential
1 und 2 entstehen, wenn eine Potentialdifferenz angelegt wird; 3 und 4 verursachen das Durchstanzen und Sedimentieren kolloidaler Partikel das Auftreten einer Potentialdifferenz.
Elektroosmose ist die Bewegung eines Dispersionsmediums relativ zu einer stationären dispergierten Phase unter dem Einfluss eines elektrischen Stroms.
Elektrophorese – Dies ist die Bewegung dispergierter Phasenpartikel relativ zu einem stationären Dispersionsmedium unter dem Einfluss eines elektrischen Stroms.
P  Der Grund für das Auftreten elektrokinetischer Phänomene ist die räumliche Trennung von Ladungen und das Auftreten einer doppelten elektrischen Schicht.
Der Grund für das Auftreten elektrokinetischer Phänomene ist die räumliche Trennung von Ladungen und das Auftreten einer doppelten elektrischen Schicht.
Die elektrische Doppelschicht ist ein Flachkondensator, eine Platte wird durch potenzialbestimmende Ionen, die andere durch Gegenionen gebildet. Die Verunreinigung der Ionen erfolgt auf die gleiche Weise, wie potenzialbestimmende Co-Ionen in das Lösungsvolumen gedrückt werden. Abstand zwischen den Platten  . Das Potenzial fällt linear ab, die Potenzialdifferenz
. Das Potenzial fällt linear ab, die Potenzialdifferenz  .
.
Eine äußere Potentialdifferenz führt zum Auftreten eines Schermoduls  ist ein Kräftepaar pro Flächeneinheit, das entlang der Oberfläche eines festen Körpers wirkt.
ist ein Kräftepaar pro Flächeneinheit, das entlang der Oberfläche eines festen Körpers wirkt.

Im Gleichgewicht ist der Schubmodul gleich dem viskosen Reibungsmodul (  ).
).

In unseren Bedingungen  ,
,

 - Gelemholtz-Smalukowski-Gleichung
- Gelemholtz-Smalukowski-Gleichung
 - lineare Geschwindigkeit der Phasenverschiebung.
- lineare Geschwindigkeit der Phasenverschiebung.
E– elektrische Feldstärke.
 - Potentialunterschied zwischen Platten
- Potentialunterschied zwischen Platten
 - Elektrophoretische Mobilität [m 2 /(V*s)].
- Elektrophoretische Mobilität [m 2 /(V*s)].
Das Helemholtz-Modell berücksichtigt nicht die thermische Bewegung von Molekülen. Tatsächlich ist die Verteilung der Ionen in der Doppelschicht komplexer.
Gui und Chapman identifizierten die folgenden Ursachen für DES:
Der Übergang eines Ions von einer Phase in eine andere, wenn ein Gleichgewicht hergestellt ist.
Ionisierung fester Phasenmaterie.
Vervollständigung der Oberfläche mit im Dispersionsmedium vorhandenen Ionen.
Polarisation von einer externen Stromquelle.
Die elektrische Doppelschicht weist eine unscharfe oder diffuse Struktur auf. Die Ionen neigen dazu, gleichmäßig in der diffusen Schicht verteilt zu sein.
Die diffuse Schicht besteht aus Kontinonen; die Länge der Schicht wird durch deren kinetische Energie bestimmt. Bei Temperaturen nahe dem absoluten Nullpunkt liegen Gegenionen möglichst nah an den potentiell bestimmenden Ionen.
Danyas Theorie basiert auf zwei Gleichungen:
Boltzmann-Gleichung

 - den Kräften der elektrostatischen Wechselwirkung entgegenwirken.
- den Kräften der elektrostatischen Wechselwirkung entgegenwirken.
 - volumetrische Ladungsdichte.
- volumetrische Ladungsdichte.

Poisson-Gleichung 
Da die Dicke der EDL viel kleiner ist als die Partikelgröße und bei einer flachen EDL die Ableitung nach Koordinaten  Und
Und  wird abgeschafft.
wird abgeschafft. 
Für e y um y<<1 функцию можно разложить в ряд Маклорена:

Beschränken wir uns also auf zwei Begriffe der Reihe:


 - Die DEL-Dicke ist der Abstand, in dem das DEL-Potenzial abnimmt e einmal.
- Die DEL-Dicke ist der Abstand, in dem das DEL-Potenzial abnimmt e einmal.
Je niedriger die Temperatur, desto weniger  . Bei T→0 – flaches DEL. Je höher die Konzentration, desto mehr ich, desto weniger
. Bei T→0 – flaches DEL. Je höher die Konzentration, desto mehr ich, desto weniger  .
.





 „–“ bedeutet, dass das Potenzial mit zunehmender Entfernung abnimmt. =>
„–“ bedeutet, dass das Potenzial mit zunehmender Entfernung abnimmt. =>
=>

 ,
,
 - Das Potenzial nimmt exponentiell ab.
- Das Potenzial nimmt exponentiell ab.
Potenzial für Oberflächenladungsdichte: 
Die Oberflächenladung ist eine Volumenladung mit umgekehrtem Vorzeichen, integriert über die Distanz.




=>


Wo das Potenzial um das 2,7-fache abnimmt - 
Doppelschichtige Kapazität 
Der Nachteil der Theorie besteht darin, dass das Vorhandensein der Helemholtz-Schicht nicht berücksichtigt wird, d. h. nicht berücksichtigt  , daher die Fehler bei der Bestimmung der Hauptparameter. Es erklärt auch nicht den Einfluss von Ionen unterschiedlicher Natur auf die Dicke der elektrischen Doppelschicht.
, daher die Fehler bei der Bestimmung der Hauptparameter. Es erklärt auch nicht den Einfluss von Ionen unterschiedlicher Natur auf die Dicke der elektrischen Doppelschicht.
Sterns Theorie. Struktur einer kolloidalen Mizelle.
Die elektrische Doppelschicht besteht aus zwei Teilen: dicht und diffus. Durch die Wechselwirkung potentiell bildender Ionen mit gezielt adsorbierten Ionen entsteht eine dichte Schicht. Diese Ionen sind in der Regel teilweise oder vollständig dehydriert und können sowohl die gleiche als auch die entgegengesetzte Ladung zu den potentiell bestimmenden Ionen haben. Sie hängt vom Verhältnis der elektrostatischen Wechselwirkungsenergie ab  und spezifisches Adsorptionspotential
und spezifisches Adsorptionspotential  . Die Ionen der dichten Schicht werden fixiert. Der andere Teil der Ionen befindet sich in der diffusen Schicht; diese Ionen sind frei und können tiefer in die Lösung eindringen, d. h. von einem Bereich höherer Konzentration zu einem Bereich niedrigerer Konzentration. Die Gesamtladungsdichte besteht aus zwei Teilen.
. Die Ionen der dichten Schicht werden fixiert. Der andere Teil der Ionen befindet sich in der diffusen Schicht; diese Ionen sind frei und können tiefer in die Lösung eindringen, d. h. von einem Bereich höherer Konzentration zu einem Bereich niedrigerer Konzentration. Die Gesamtladungsdichte besteht aus zwei Teilen.

 -Ladung der Helmholtz-Schicht
-Ladung der Helmholtz-Schicht
 -Diffuse Schichtladung
-Diffuse Schichtladung
Die Oberfläche verfügt über eine bestimmte Anzahl von Adsorptionszentren, von denen jedes mit einem Gegenion wechselwirkt. Die Konstante einer solchen quasichemischen Reaktion ist gleich:

 , Wo
, Wo  - Molenbruch der Gegenionen in Lösung
- Molenbruch der Gegenionen in Lösung
Helmholtz-Verteilung
Das Potenzial nimmt linear ab
Gouy-Potenzialverteilung. Es gibt keine dichte Schicht, das Potenzial nimmt vom Wert exponentiell ab 
Sternverteilung.
Der potenzielle Rückgang verläuft zunächst linear und dann exponentiell.
Wenn bei der Elektrophorese ein elektrisches Feld angelegt wird, bewegt sich nicht das Teilchen der festen Phase direkt, sondern das Teilchen der festen Phase mit einer ihn umgebenden Ionenschicht. Der DES wiederholt die Form des Partikels der dispergierten Phase. Beim Anlegen einer Spannung wird ein Teil der diffusen Schicht abgerissen. Die Bruchlinie heißt gleitende Grenze.
Das Potential, das an der Gleitgrenze infolge der Ablösung eines Teils der diffusen Schicht entsteht, wird aufgerufen elektrokinetisches Potenzial(Zetapotential  ).
).
Als Teilchen wird eine dispergierte Phase mit einer umgebenden Schicht aus Gegenionen und einer doppelten elektrischen Schicht bezeichnet Mizelle.
Regeln zum Schreiben kolloidaler Mizellen:


1-1 Ladeelektrolyt
T – Partikel in dispergierter Phase.
AA ist die Grenze zwischen den dichten und diffusen Teilen.
BB – Gleitgrenze.
Die Gleitgrenze kann mit der Linie AA zusammenfallen oder auch nicht.
Man nennt den pH-Wert, bei dem das Zetapotential Null ist isoelektrischer Punkt.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. Überschüssiges CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 X + X Cl - - Mizellen-Notation.
CaSO 4 m – Zuschlagstoff.
CaSO 4 m∙nCa 2+ – Kern.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - Teilchen.
2. Überschüssiges Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - Mizelle
CaSO 4 m – Zuschlagstoff.
CaSO 4 m∙nSO 4 2 + – Kern.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - Teilchen
Gelemholtz-Smoluchowski-Gleichung

 - lineare Geschwindigkeit der Grenzverschiebung (bei Elektroosmose).
- lineare Geschwindigkeit der Grenzverschiebung (bei Elektroosmose).
 - Potentialdifferenz zwischen den Kondensatorplatten (bei Elektroosmose).
- Potentialdifferenz zwischen den Kondensatorplatten (bei Elektroosmose).


 - Volumenstrom der Lösung, S– Querschnittsfläche der Zelle.
- Volumenstrom der Lösung, S– Querschnittsfläche der Zelle.

E– elektrische Feldstärke.


 (für Elektroosmose).
(für Elektroosmose).
Für Strömungspotential:

 - Potenzial
- Potenzial
 - Druck auf die Membran
- Druck auf die Membran
In der Regel sind die Werte der elektrophoretischen Mobilitäten und elektroosmotischen Mobilitäten geringer als die berechneten. Dies geschieht aus folgenden Gründen:
Entspannungseffekt (wenn sich ein Teilchen einer dispergierten Phase bewegt, wird die Symmetrie der ionischen Atmosphäre gebrochen).
Elektrophoretische Hemmung (das Auftreten zusätzlicher Reibung durch die Bewegung von Gegenionen).
Verzerrung der Stromlinien bei elektrisch leitfähigen Partikeln.
Zusammenhang zwischen Oberflächenspannung und Potenzial. Lippmann-Gleichung.
Die Bildung von EDL erfolgt spontan aufgrund des Wunsches des Systems, seine Oberflächenenergie zu reduzieren. Unter konstanten Bedingungen T Und P Die verallgemeinerte Gleichung des ersten und zweiten Hauptsatzes der Thermodynamik sieht wie folgt aus:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. Lippmann-Gleichung.
- 1. Lippmann-Gleichung.
 - Oberflächenladungsdichte.
- Oberflächenladungsdichte.
 - Differenzkapazität.
- Differenzkapazität.
 - 2. Lippmann-Gleichung.
- 2. Lippmann-Gleichung.
MIT– Kapazität.
Lösen wir die 1. Lippmann-Gleichung und die grundlegende Adsorptionsgleichung:
 ,
,


 , Dann
, Dann
 - Nernst-Gleichung
- Nernst-Gleichung
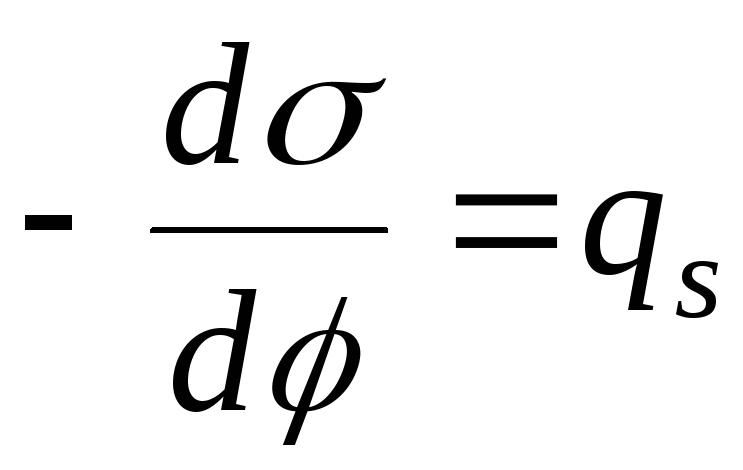 ,
,
 ,
,




 - Gleichung der Elektrokapillarkurve (ECC).
- Gleichung der Elektrokapillarkurve (ECC).
IN  :
: , Aber
, Aber 
Kationische Tenside (CPAS) reduzieren den kathodischen Zweig des EKC.
Anionische Tenside (APS) reduzieren den anodischen Zweig des EKC.
Nichtionische Tenside (NSAS) reduzieren den mittleren Teil des ECC.
Stabilität verteilter Systeme. Trennender Druck.
Verteilte Systeme können unterteilt werden:
Thermodynamisch instabile Systeme können aufgrund des Übergangs in einen metastabilen Zustand kinetisch stabil sein.
Es gibt zwei Arten von Stabilität:
Sedimentationsstabilität (relativ zur Schwerkraft).
Aggregative Stabilität. (relativ zur Adhäsion)
Koagulation ist ein Prozess der Partikeladhäsion, der zum Verlust der Aggregationsstabilität führt. Die Koagulation kann durch Temperaturänderungen, pH-Wert, Rühren und Ultraschall verursacht werden.
Bei der Koagulation wird unterschieden:
Reversibel.
Irreversibel.
Die Koagulation erfolgt unter Einführung von Elektrolyten.
Koagulationsregeln:

Film- Dies ist der Teil des Systems, der zwischen zwei Grenzflächen liegt.
Trennender Druck tritt auf, wenn die Filmdicke durch die Wechselwirkung sich nähernder Oberflächenschichten stark abnimmt.

„-“ – mit abnehmender Filmdicke steigt der Trenndruck.
P 0 ist der Druck in der Volumenphase, die eine Fortsetzung der Zwischenschicht darstellt.
P 1 – Druck im Film.
Theorie der Stabilität. DLFO (Deryagin, Landau, Fairway, Overbeck).
Nach der DLFO-Theorie besteht der Disjunktionsdruck aus zwei Komponenten:
Elektrostatisch P E (positiv, es liegt an den Kräften der elektrostatischen Abstoßung). Entspricht einer Abnahme der Gibbs-Energie mit zunehmender Filmdicke.
Molekular P M (negativ, aufgrund der Wirkung anziehender Kräfte). Sie entsteht durch Filmkompression aufgrund chemischer Oberflächenkräfte, der Wirkungsradius der Kräfte beträgt Zehntel nm mit einer Energie von etwa 400 kJ/mol.
Gesamte Interaktionsenergie:

 - Das System ist insgesamt stabil
- Das System ist insgesamt stabil
 - instabiles System
- instabiles System
P  positive Komponente.
positive Komponente.
Der Anstieg ist auf einen Anstieg der potentiellen Energie beim Komprimieren dünner Filme zurückzuführen. Bei Filmen großer Dicke wird die überschüssige Ionenenergie kompensiert und entspricht der Energiewechselwirkung im Volumen des Dispersionsmediums.
Wenn  (
( - Schichtdicke,
- Schichtdicke,  (Ionenradius) führt eine Verdünnung des Films zum Verschwinden und zur Reduzierung von darin enthaltenen Molekülen und Ionen mit minimaler Oberflächenenergie. Die Anzahl benachbarter Teilchen nimmt ab, wodurch die potentielle Energie der im Film verbleibenden Teilchen zunimmt.
(Ionenradius) führt eine Verdünnung des Films zum Verschwinden und zur Reduzierung von darin enthaltenen Molekülen und Ionen mit minimaler Oberflächenenergie. Die Anzahl benachbarter Teilchen nimmt ab, wodurch die potentielle Energie der im Film verbleibenden Teilchen zunimmt.


Die DLVO-Theorie betrachtet die Wechselwirkung von Teilchen als die Wechselwirkung von Platten.

Teilchen interagieren nicht



 - Laplace-Gleichung,
- Laplace-Gleichung,  ,
,


Für schwach geladene Oberflächen
Für stark aufgeladene Oberflächen:

Die molekulare Komponente ist die Wechselwirkung zweier Atome:
 ~
~
Wechselwirkung eines Atoms mit einer Oberfläche:

Nehmen wir zwei Datensätze:
D  Um die molekulare Komponente zu erhalten, müssen alle Wechselwirkungsenergien der Atome der rechten und linken Platte summiert werden.
Um die molekulare Komponente zu erhalten, müssen alle Wechselwirkungsenergien der Atome der rechten und linken Platte summiert werden.
Wo  - Hamaker-Konstante (berücksichtigt die Art der interagierenden Körper).
- Hamaker-Konstante (berücksichtigt die Art der interagierenden Körper).
Das. Die Wechselwirkungsenergie von Teilchen in einem System kann mithilfe von Potentialkurven ausgedrückt werden.
I – Primärpotentialminimum. Dies ist eine Zone irreversibler Koagulation, die Anziehungskräfte überwiegen.
II – Zone aggregierter Stabilität, abstoßende Kräfte überwiegen.
III – sekundäres Potentialminimum (oder Flockungszone). Zwischen den Partikeln der dispergierten Phase befindet sich eine Elektrolytschicht, und die Partikel können getrennt und in die Zone der Aggregationsstabilität überführt werden.

Kurve 1 – das System ist insgesamt stabil.
Kurve 2 – stabil in Zone I, instabil in Zone II.
Kurve 3 – im System ist eine Koagulation aufgetreten.
Kurve 4 – am Punkt 4 ist die gesamte Wechselwirkungsenergie U=0,  Dieser Extrempunkt entspricht dem Beginn einer schnellen Koagulation.
Dieser Extrempunkt entspricht dem Beginn einer schnellen Koagulation.
Es gibt zwei Fälle:
1. Leicht aufgeladene Oberflächen:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - Dies ist die Schichtdicke, die dem Beginn des Koagulationsprozesses entspricht.
- Dies ist die Schichtdicke, die dem Beginn des Koagulationsprozesses entspricht.






 - für schwach geladene Oberflächen
- für schwach geladene Oberflächen
 Dann
Dann 
2. Für stark geladene Oberflächen:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Lass uns quadrieren (3)

Koagulation:
Bei der spezifischen Adsorption können Ionen in überäquivalenten Mengen adsorbiert werden, sodass die Oberfläche ihre Ladung ändern kann. Die Oberfläche wird wieder aufgeladen.
Bei der spezifischen Adsorption können nicht nur Ionen mit entgegengesetztem Vorzeichen, sondern auch mit gleichem Vorzeichen adsorbiert werden.
Wenn Ionen mit dem gleichen Vorzeichen wie die Oberfläche adsorbiert werden, kommt es in der Oberflächenschicht nicht zu einem Abfall des Potentials, sondern zu einem Anstieg.

Neutralisationskoagulation (tritt unter Beteiligung schwach geladener Teilchen auf und hängt nicht nur von der Ladung des Elektrolytkoagulators, sondern auch vom Potential an der Grenze der dichten und diffusen Schichten ab).
Smoluchowskis Theorie der schnellen Gerinnung.

Abhängigkeit der Gerinnungsgeschwindigkeit von der Elektrolytkonzentration.
I – Gerinnungsrate ist niedrig,
II – die Koagulationsrate ist nahezu proportional zur Elektrolytkonzentration.
III – Bereich der schnellen Koagulation, die Geschwindigkeit ist praktisch unabhängig von der Konzentration.
Grundbestimmungen:
Das Ausgangssol ist monodispers, ähnliche Partikel haben eine Kugelform.
Alle Teilchenkollisionen sind wirksam.
Wenn zwei Primärteilchen kollidieren, entsteht ein Sekundärteilchen. Sekundär + Primär = Tertiär. Primär, sekundär, tertiär – Vielfalt.
In Bezug auf die chemische Kinetik kann der Koagulationsprozess durch die Gleichung beschrieben werden:
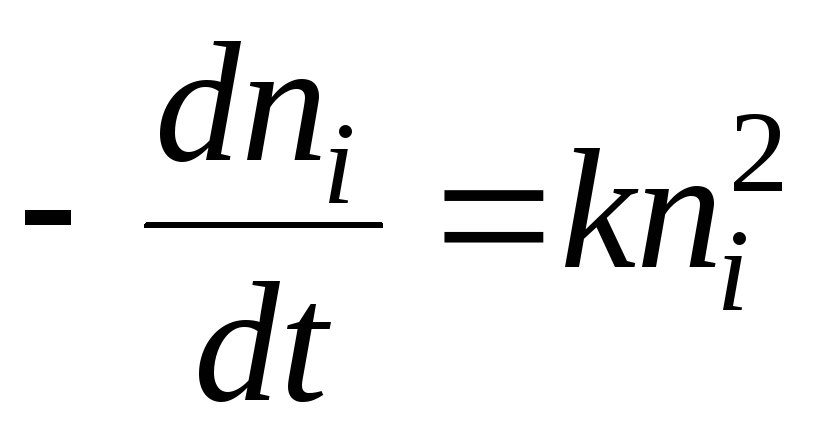
Die Lösung wird die Gleichung sein: 
 - halbe Gerinnungszeit. Dies ist die Zeit, in der die Anzahl der Solpartikel um das Zweifache abnimmt.
- halbe Gerinnungszeit. Dies ist die Zeit, in der die Anzahl der Solpartikel um das Zweifache abnimmt.

 ,
,
 ,
,
 ,
,


Mit zunehmender Multiplizität verschiebt sich das Maximum der Gerinnungskurven zu größeren Werten  .
.
Mängel:
Annahme der Monodispersität.
Annahme über die Wirksamkeit aller Kollisionen.


