Адсорбція має місце межі розділу фаз. Тому термодинамічний опис поверхневих явищ є доцільним розглядати як окремий випадок термодинаміки гетерогенних систем.
Мал. 3.4. Адсорбція по Гіббсу: 1 - двофазна система порівняння, 2 - реальна двофазна система з неоднорідною областю
У термодинаміці гетерогенних систем використовується принцип адитивності, який полягає в наступному: всі екстенсивні властивості гетерогенної системи дорівнюють сумі відповідних екстенсивних властивостей, якими мали б фази до того, як їх привели в контакт.Позначимо фази через α та β (рис.4). Тоді для ідеальної системи, такої, що властивості фаз поблизу поверхні розділу збігаються з їх об'ємними властивостями, для внутрішньої енергії U, об'єму V, маси (числа молів) n, ентропії S після встановлення рівноваги в гетерогенній системі справедливі співвідношення:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
При цьому мається на увазі, що температура та тиск в обох фазах однакові.
Для реальних гетерогенних систем перехідна область межі двох фаз робить додатковий внесок у екстенсивні властивості системи. Якщо поверхневі явища мають місце, слід враховувати відмінність екстенсивних властивостей реальної гетерогенної системи екстенсивних властивостей модельної системи, в якій поверхневі явища відсутні. Така система називається системою порівняння. Система порівняння має ті ж інтенсивні параметри (T, P, C i ...) і такий же обсяг V, що і реальна система (рис. 4).
З термодинамічної точки зору під величиною адсорбції Р розуміють надмірну кількість речовини n s , виражену в молях або грамах, якою володіє реальна гетерогенна система в порівнянні з системою порівняння, віднесена до площі поверхні розділу фаз або до площі поверхні адсорбенту А. Приймається, що система порівняння має тими ж інтенсивними параметрами (T, P, Ci), і таким самим обсягом (V = V α + V β), що і реальна система (рис.4).
Г = (n - n α - n β)/A = n s /A 3.11
Надмірні термодинамічні функції перехідної області реальної системи (позначимо їх індексом s) можна записати як
U s = U - U α - U β , ns = n - n α - n β , S s = S - S α - S βі т.д.
Експериментальні вимірювання адсорбції завжди дають адсорбцію саме як надлишок компонента реальної системі порівняно з обраної системою порівняння. Наприклад, при адсорбції газу на твердому адсорбенті або при адсорбції компонентів на твердій фазі для знаходження величин адсорбції визначають зміну початкових концентрацій адсорбату після зіткнення фаз α і β
n i s = V (C i o - C i),
де C i o- вихідні концентрація i-го компонента, C i– концентрація i – го компонента після встановлення рівноваги між дотичними фазами. При цьому вважається, що обсяг Vне змінюється. Однак, концентрація i-го компонента C iотримана експериментально, визначається в обсязі V’над поверхнею розділу фаз без урахування обсягу неоднорідної області перехідного шару V αбіля межі розділу, де концентрація становить C i α. Таким чином, зважаючи на існування в реальній системі неоднорідної області, загальний обсяг системи можна представити як V = V' + V α. Вся кількість i-го компонента C i oрозподілиться між цими двома обсягами:
V C i o = V C i + V α C i α ,
і число молей компонента i, адсорбованого на поверхні розділу фаз, дорівнюватиме
n i s = (V'C i + V α C i α) - (V' + V α)C i = V α (C i α - C i) 3.12
Тобто. визначається експериментально адсорбція є надлишок i-го компонента в об'ємі V α в порівнянні з кількістю цього компонента в такому ж обсязі далеко від поверхні розділу фаз. Саме така адсорбція називається адсорбцією за Гіббсом. .
V α C i αназивається повним змістом i-го компонента в адсорбційному шарі. В області дуже малих концентрацій C iв обсязі V’поправкою V α C iрівняння (3.2) можна знехтувати та вважати виміряну величину V α C i αповним змістом i-го компонента в адсорбційному шарі, наприклад при адсорбції газу на твердому адсорбенті при низьких тисках.
Термодинаміка адсорбційних процесів.
| Найменування параметру | Значення |
| Тема статті: | Термодинаміка адсорбційних процесів. |
| Рубрика (тематична категорія) | Освіта |
Основні визначення та способи класифікації адсорбційних процесів.
Адсорбція відноситься до явищ, що відбуваються внаслідок мимовільного зменшення поверхневої енергії.
Адсорбція– процес мимовільного оборотного або незворотного перерозподілу компонентів гетерогенної системи між поверхневим шаром та об'ємом гомогенної фази.
У багатокомпонентних системах поверхневий шар переважно переходить компонент, який сильніше знижує міжфазний натяг. В однокомпонентних системах при формуванні поверхневого шару відбувається зміна його структури (певна орієнтація атомів і молекул, поляризація), яка називається автоадсорбцією.
Більш щільну фазу, на якій локалізовані адсорбційні взаємодії, називають адсорбентом. Речовина, що перерозподіляється між об'ємом гомогенної фази і поверхневим шаром, позначають терміном адсорбатʼʼ.
У ряді випадків процес адсорбції є оборотним. У цьому випадку за певних умов частина адсорбованих молекул в результаті молекулярно-кінетичних явищ може перейти з поверхневого шару в об'єм фази. Процес, зворотний адсорбції, називають десорбцією.
Методи класифікації адсорбційних процесів.
Класифікація адсорбційних процесів за агрегатним станом взаємодіючих фаз.Враховуючи залежність отагрегатного стану суміжних фаз розрізняють такі типи адсорбційних процесів:
Адсорбція газів на твердих адсорбентах;
Адсорбція розчинених речовин на межах розділу «тверде тіло – рідина» і «рідина – рідина»;
Адсорбція поверхнево-активних речовин на межі розділу «рідина – газ».
Класифікація адсорбційних процесів за механізмом взаємодії адсорбенту та адсорбату.Адсорбцію можна розглядати як взаємодію молекул адсорбату з активними центрами адсорбенту. За механізмом їхньої взаємодії поділяють такі види адсорбції:
1) фізична (молекулярна) адсорбція– взаємодія між молекулами адсорбату та адсорбенту здійснюється за рахунок сил Ван-дер-Ваальса, водневих зв'язків (без протікання хімічних реакцій);
2) хімічна адсорбція (хемосорбція)– приєднання молекул адсорбату до активних центрів адсорбенту відбувається в результаті протікання хімічних реакцій різних типів (за винятком реакцій іонного обміну);
3) іонообмінна адсорбція (іонний обмін) – перерозподіл речовини адсорбату між розчином і твердою фазою (іонітом) за механізмом реакцій іонного обміну.
Для кількісного опису адсорбційних процесів застосовують дві величини.
1) Абсолютна адсорбція- Кількість (моль) або маса (кг) адсорбату на одиницю площі поверхні або маси адсорбенту. Позначення – А; розмірність: моль/м 2 , моль/кг, кг/ м 2 , кг/кг.
2) Гіббсівська (надлишкова) адсорбція- надлишок речовини адсорбату в поверхневому шарі певної товщини в порівнянні з його кількістю в об'ємі гомогенної фази, віднесений до одиниці площі поверхні або маси адсорбенту. Позначення – Р; розмірність: моль/м 2 , моль/кг.
Зв'язок між абсолютною та надмірною адсорбцією можна проілюструвати за допомогою рівняння:
Г = А - з * h (3.1)
де с - рівноважна концентрація речовини в об'ємі фази, моль/м 3;
h - товщина поверхневого шару, що умовно приймається рівною 10 -9 м.
У багатокомпонентних гетерогенних системах при перерозподілі того чи іншого компонента між об'ємом гомогенної фази і поверхневим шаром справедливе рівняння для надлишкової внутрішньої енергії поверхні:
U = T * S + s * s + Sm i * ni (3.2)
Привівши всі члени рівняння до одиниці площі міжфазної поверхні, отримаємо:
U s = T * S s + s + Sm i * Г i (3.3)
де Г i = n i / s - надлишок i-го компонента в поверхневому шарі, тобто гіббсівська адсорбція.
Для однокомпонентної системи рівняння (3.3) набуде вигляду:
G s = s + m * Г (3.4)
де G s = U s - T * S s - Енергія Гіббса поверхні або робота створення одиниці площі поверхні;
m * Г – ущільнення речовини речовини, що адсорбується в поверхневому шарі.
Виходячи з рівняння (3.4) можна зробити висновок про те, що при адсорбції робота зі створення міжфазної поверхні складається з роботи утворення поверхні (розриву когезійних зв'язків в об'ємі фази адсорбату) та ущільнення речовини у поверхневому шарі.
У стані динамічної рівноваги між адсорбентом і адсорбатом зміна енергії Гіббса гетерогенної системи ΔG = 0, термодинаміка процесу адсорбції описується рівнянням, що отримало назву фундаментальне адсорбційне рівняння Гіббса:
Ds = SГ i * dm i (3.5)
Дане рівняння є універсальним, оскільки справедливо для всіх типів адсорбційних процесів
Окремі випадки адсорбційного рівняння Гіббса.
1) Адсорбція із розчинів.
Для хімічного потенціалу i-го компонента системи при протіканні адсорбції на межах розділу «рідина – твердий адсорбент» і «рідина – газ» справедливі рівняння:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
де m i 0 - Хімічний потенціал i -го компонента системи за стандартних умов;
a i – активність i компонента системи за стандартних умов.
Виходячи з цього, адсорбційне рівняння Гіббса набуде вигляду:
Г i = - a i / R * T * (ds / da i) (3.8)
Для розчинів неелектролітів приймаємо a i = с i тоді:
Г i = - з / R * T * (ds / dс) (3.9)
Для розчинів електролітів:
Г i = - з ± n / R * T * (ds / dс ± n) (3.10)
де з ± – середня іонна концентрація розчину;
n – стехіометричний коефіцієнт.
2) Адсорбція речовин із газової фази.
Відповідно до рівняння Менделєєва-Клайперона:
Р = з * R * T (3.11)
У зв'язку з цим рівняння Гіббса для адсорбції газів на твердих адсорбентах записують у наступній формі:
Г i = - Р/R*T* (ds/dР) (3.12)
На практиці адсорбційне рівняння Гіббса дозволяє за даними вимірювання поверхневого натягу при різних значеннях концентрації рідини або рівноважного тиску газу розрахувати величину адсорбції речовин у міжфазному шарі, для якого визначено поверхневий натяг.
Термодинаміка адсорбційних процесів. - Поняття та види. Класифікація та особливості категорії "Термодинаміка адсорбційних процесів." 2017, 2018.
Адсорбція як мимовільне концентрування молекул лежить на поверхні супроводжується зниженням ентропії системи. Оскільки критерієм мимовільності процесу є
∆Н - T · ∆S = ∆G< 0,
то адсорбція можлива лише при ∆Н< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. При підвищенні температури рівновага зміщується у бік ендотермічного процесу, тобто десорбції.
Адсорбція на поверхні твердого тіла
1. Мономолекулярна адсорбція.
По теорії Ленгмюра молекули адсорбтиву взаємодіють із поверхнею адсорбенту, утворюючи в результаті мономолекулярний шар. В цьому випадку ступінь заповнення () поверхні адсорбується при адсорбції з газової фази
з рідини
де К - константа рівноваги (константа адсорбції);
р - парціальний тиск газу, що адсорбується;
с - концентрація речовини, що адсорбується.
Залежність від р (або с) представлена графіком (ізотерма адсорбції, Т = const) на рис. 1.3.

Мал. 1.3. Ступінь заповнення поверхні речовиною, що адсорбується.
При малих концентраціях та парціальних тисках адсорбція пропорційна концентрації або парціальному тиску:
р<< 1, β ≈ К· р абос<< 1, β ≈ К· з, тобто. початкова ділянка ізотерми приблизно лінійна, причому tg α = К(tg α визначають по нахилу кривої при р (або с) → 0: або ).
Якщо кількість молей адсорбованої речовини на 1 г адсорбенту; - максимально можливу кількість молей адсорбованої речовини на 1 г адсорбенту ("ємність моношару"), то
Підставляючи β у рівняння (1.3) (для випадку адсорбції з газової фази концентрацію зу рівняннях слід замінити на тиск р), отримуємо:
 (1.6)
(1.6)
Так як і К у цій парі адсорбент-адсорбтив є константами (при T=const), то залежно можна знайти і До(Рис. 1.4).

Мал. 1.4. Графічне рішення рівняння адсорбції
одержують шляхом екстраполяції експериментальної лінійної залежності до () = 0; і, оскільки , то , .
Величину можна використовувати для визначення питомої поверхні адсорбенту УД (м 2 на 1 г адсорбенту), якщо відома площа ω, займана на поверхні однією молекулою адсорбтиву (визначається з розмірів молекули):
УД = · ω · Nа, (1.7)
де Nа – число Авогадро (Nа = 6,02 · 10 23).
У свою чергу, відому величину УД можна використовувати для розрахунку або будь-якої речовини з його адсорбції на даному адсорбенті.
2. Полімолекулярна адсорбція.
Рівняння (1.5) визначає криву з насиченням, тобто. при
р (або с) → ∞ прагне граничного значення, що дорівнює (рис. 1.5,а).

Рис.1.5. Ізотерми адсорбції:
а – адсорбція з насиченням; б – полімолекулярна адсорбція
Однак у деяких випадках ізотерми адсорбції виглядають як показано на рис. 1.5, б, тобто. не досягає межі навіть за високих р (або с).
Залежність типу показаної на рис. 1.5 б відповідають полімолекулярної адсорбції. Як правило, такі ізотерми характерні для речовин із сильними міжмолекулярними взаємодіями (наприклад, для води). Коли центри адсорбції на поверхні адсорбенту зайняті (мономолекулярний шар насичений), "посадка" наступних молекул адсорбату відбувається за рахунок міжмолекулярних взаємодій з адсорбованими молекулами (рис.1.6). Теплота такої адсорбції близька за абсолютною величиною, але протилежна за знаком теплоти випаровування відповідної рідини (подумайте, чому).

Рис.1.6. Схема адсорбції:
а – мономолекулярна адсорбція; б - полімолекулярна адсорбція
У міру наближення рдо тиску насиченої пари адсорбованої речовини воно починає конденсуватися на поверхні адсорбенту, в результаті швидко росте зі зростанням р.
У разі взаємодії двох атомів:
U – енергія взаємодії;
U = U ПРИТЯЖ. + U ВІДТАЛ.
 - рівняння Леннарда-Джонса
, c, b, m = const
- рівняння Леннарда-Джонса
, c, b, m = const
У випадках взаємодії атомів з твердою поверхнею необхідно провести підсумовування всіх взаємодій.
х-відстань до поверхні
r – радіус дії сил тяжіння
dV – обсяг
n – число молекул поверхні
U АДС. – енергія адсорбційної взаємодії



У разі адсорбції тяжіння посилюється. І у разі при взаємодії типу неполярно-неполярна адсорбція переважно локалізується у поглибленнях.
Електростатична взаємодія.
Полярний адсорбент – неполярний адсорбат
Неполярний адсорбент – полярний адсорбат
Полярний адсорбент – полярний адсорбат.
М  олекулу адсорбату представляють як диполь, а адсорбенту – як провідник, у якому молекула адсорбату індукує диполь дзеркально симетрично стосовно даного.
олекулу адсорбату представляють як диполь, а адсорбенту – як провідник, у якому молекула адсорбату індукує диполь дзеркально симетрично стосовно даного.
X – відстань до середини
При взаємодії виникає потенціал:
 ,
,
 - Дипольний момент.
- Дипольний момент.
Потенціал прагне набути максимального значення, тобто. диполі прагнуть зорієнтуватися перпендикулярно поверхні.
Оскільки підвищення температури сприяє зростанню броунівського руху, воно призводить до гальмування процесу адсорбції.
У разі електростатичної взаємодії адсорбат переважно локалізується на виступах.
Фундаментальне адсорбційне рівняння.
У разі адсорбції відбувається перерозподіл компонента, отже, змінюється хімічний потенціал. Процес адсорбції можна як перехід поверхневої енергії в хімічну.
Об'єм шару = 0, тоді узагальнене рівняння I та II закону термодинаміки:
T = const; (1) = (2) => 

Для двокомпонентної системи:
,
 ,
,
 =>
=>
=>
 - адсорбційне рівняння Гіббса
.
- адсорбційне рівняння Гіббса
.
Для випадку адсорбції тб. тіло – газ: , 
 ,
,

 - ізотерма
- ізотерма
 - ізобара
- ізобара
 - Ізопікна
- Ізопікна
 - ізостера
- ізостера
Ізотерма, ізопікна, ізостера пов'язані один з одним.
Т.к. адсорбція функція 



Ізотерма Генрі Ізотерма Лангмюра



Термодинаміка. Адсорбція.
Для конденсованих середовищ:
 ,
,
 ,
,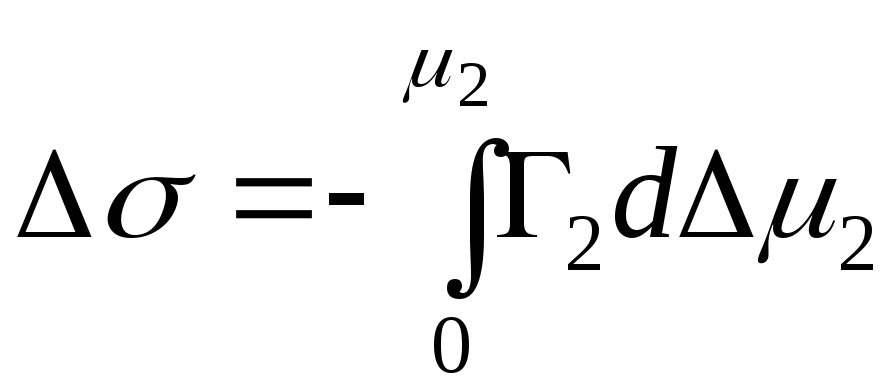
 - інтегральна зміна енергії Гіббса
.
- інтегральна зміна енергії Гіббса
.

P–тиск над викривленою поверхнею, P S–тиск над плоскою поверхнею
 - адсорбційний потенціал
- адсорбційний потенціал
Диференційна зміна ентрапії 
 , Г = const
, Г = const
- диференційна зміна ентропії
- диференціальна ентальпія адсорбції
 - ізостерична теплота адсорбції
- ізостерична теплота адсорбції
 - теплота конденсації
- теплота конденсації
 - чиста теплота адсорбції
- чиста теплота адсорбції

 ,
,




Qa – інтегральна теплота адсорбції, 
Qra – інтегральна чиста теплота адсорбції,

Рівняння Генрі
Дослідження адсорбції не може бути неоднорідністю поверхні, тому найпростіші закономірності отримують для однорідних поверхонь.
Розглянемо взаємодію газів з твердою поверхнею, коли здійснюється перехід газу з рівноважного стану в обсязі рівноважний стан на поверхні. Цей випадок аналогічний до рівноваги газів у полі сили тяжіння.
 ,
,
 ,
=>
,
=> -рівняння Генрі
-рівняння Генрі
 - Коефіцієнт розподілу
- Коефіцієнт розподілу
У процесі адсорбції відбувається зміна хімічних потенціалів.
Для об'ємної фази: 
Для газу на поверхні: 
У стані рівноваги  , тобто.
, тобто.

У рівнянні Генрі константа залежить від концентрації
Рівняння Генрі виконується в області низьких тисків та концентрацій. У міру зростання концентрації можливі 2 типи відхилень від закону Генрі:
1 - позитивні відхилення, D зменшується, А зменшується
2 – негативні відхилення, D – збільшується, А – збільшується.
Тип відхилення визначається переважанням тієї чи іншої типу взаємодії адсорбент-адсорбат.
При сильному адгезійному взаємодії коефіцієнти активності зростають – позитивне відхилення. У разі когезійних взаємодій спостерігаються негативні відхилення.
Мономолекулрова адсорбція.
Ізотерма Лангмюр.
Найпростіші закономірності були отримані теоретично генрі. Ленгмюр запропонував теорію, за якою, адсорбція сприймається як квазихимическая реакція. При цьому:
Поверхня енергетично однорідна.
Адсорбція локалізована, кожен адсорбційний центр взаємодіє з однією молекулою адсорбату.
Молекули адсорбату не взаємодіють одна з одною.
Адсорбція моношарова.

 - Поверхня,
- Поверхня,  - адсорбат,
- адсорбат,  - Адсорбційний комплекс.
- Адсорбційний комплекс.
 тоді концентрація адсорбційних місць:
тоді концентрація адсорбційних місць:  ,
, - Гранична адсорбція.
- Гранична адсорбція.
 тоді константа реакції:
тоді константа реакції: 

 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Залежність адсорбції від концентрації
1 )
)
 ,
,
2) область високих концентрацій
 - гранична адсорбція, утворення мономолекулярного шару
- гранична адсорбція, утворення мономолекулярного шару
Для енергії Гіббса: .
g – ентропійний множник.
У разі ізотерми Генрі енергія Гіббса характеризує перехід адсорбату зі стандартного стану в об'ємі стандартний стан на поверхні. У разі ізотерми Ленгмюра  характеризує ступінь спорідненості адсорбенту та адсорбату.
характеризує ступінь спорідненості адсорбенту та адсорбату.
 знаходять із ізобари Вант-Гоффа.
знаходять із ізобари Вант-Гоффа.

 тоді
тоді  , звідси
, звідси  .
.

 - Ступінь заповнення поверхні.
- Ступінь заповнення поверхні.




 - кількість вільних місць,
- кількість вільних місць,  - Число зайнятих місць.
- Число зайнятих місць.
 ,
,

Тобто. в області високих концентрацій кількість вільних місць обернено пропорційно кількості адсорбату.
Адсорбція суміші газів на однорідній поверхні.
У цьому випадку процес адсорбції розглядають як дві паралельно протікають реакції.
 (1)
(1)

 (2)
(2)


Адсорбція суміші газів на неоднорідній поверхні.
У разі неоднорідної поверхні не можна обмежуватись середніми заповненнями.
Внаслідок конкурентної боротьби на ділянках різних типів можлива локалізація різних адсорбатів.
У цьому випадку відношення  .
.
 ,
,
 - Тиск насиченої пари адсорбату.
- Тиск насиченої пари адсорбату.
 ,
,
 – теплоти адсорбції.
– теплоти адсорбції.
"+" - симбатна залежність, "-" - антибатна залежність, "Н" - кореляції немає.
«+» - адсорбція протікає за однаковим механізмом. На найбільш енергетично вигідних ділянках переважно адсорбується газ, що має велику спорідненість до поверхні.
«-» - адсорбція протікає за різними механізмами і до певного моменту часу конкурентної боротьби за поверхню немає.
Мономолекулярна адсорбція переважно реалізується при фізичній адсорбції газів за малих значень p, а також на межі розділу рідина/газ.
Полімолекулярна адсорбція.
Теорія БЕТ(Брунауер, Еммет, Теллер).
У разі коли утворення моношару недостатньо для компенсації поверхневої енергії, адсорбція полімолекулярна і її можна розглядати як результат вимушеної конденсації під дією поверхневих сил.
Основні положення:
У разі потрапляння молекули адсорбату на зайняте місце утворюється кратний комплект.
У міру наближення pдо p sзменшується кількість вільних адсорбційних місць. Спочатку збільшується, та був зменшується кількість місць, зайнятих одиничними, подвійними тощо. комплектами.
При p =p s адсорбція перетворюється на конденсацію.
Горизонтальні взаємодії відсутні.
Для першого шару виконується ізотерма Лангмюр.
Поверхня сприймається як сукупність адсорбційних місць. Справедлива умова динамічної рівноваги: швидкість конденсації на вільних місцях дорівнює швидкості випаровування із зайнятих.
a – коефіцієнт конденсації (частка молекул, що сконденсувалися на поверхні);
 ,
,
Zm – максимальна кількість вільних місць.
 - Частота коливань атомів у напрямку перпендикулярному до поверхні.
- Частота коливань атомів у напрямку перпендикулярному до поверхні.
Для першого шару умови динамічної рівноваги:



 тоді
тоді
 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Для другого шару буде справедливо: 
Для i-го шару: 
Для спрощення приймають, що і ν однакові всім шарів, крім першого. Для всіх шарів, крім першого теплота адсорбції, постійна. Для останнього шару теплота адсорбції дорівнює теплоті конденсації. В результаті було отримано рівняння
 (*)
(*)
C- Константа,
У разі теорії БЕТ константа Зхарактеризує енергію Гіббса чистої адсорбції. Рівняння містить лише одну константу, а також це рівняння дуже важливе для визначення питомої поверхні адсорбенту.
Оскільки внаслідок адсорбції теплота виділяється, визначення питомих поверхонь ведуть за низьких температур.
 ????????????
????????????


Основний недолік теорії- Нехтування горизонтальними взаємодіями на користь вертикальних.
Рівняння виконується в області значень  від 0,05 до 0,3.
від 0,05 до 0,3.
Там де  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – позначається взаємодія адсорбат – адсорбат.
> 0,3 – позначається взаємодія адсорбат – адсорбат.
Облік взаємодій адсорбат-адсорбат.
Взаємодія виявляється при адсорбції на неполярній поверхні розгалужених молекул або молекул. Здібних утворювати асоціати. І тут змінюється форма ізотерм адсорбції.
А  дсорбент не полярний.
дсорбент не полярний.
Графіку 1 відповідають слабкі взаємодії адсорбат-адсорбат, сильне адсорбат-адсорбент.
Графіку 2 відповідають сильне взаємодія адсорбат-адсорбат, сильне адсорбат-адсорбент.
Графіку 3 відповідають сильну взаємодію адсорбат-адсорбат, слабку адсорбат-адсорбент.
 ,
,

У разі взаємодії між молекулами адсорбату потрібний облік зміни коефіцієнтів активності. І це рівняння записують у вигляді:
 - Рівняння Фрункіна, Фаулера, Гугенгейма.
- Рівняння Фрункіна, Фаулера, Гугенгейма.
k- Атракціонна постійна.
Потенційна теорія Поляні.
Ця теорія не виводить будь-якого типу ізотерми адсорбції, а дозволяє розрахунку ізотерм при іншій температурі.
Адсорбція– це результат тяжіння адсорбату до поверхні адсорбенту за рахунок дії адсорбційного потенціалу, який не залежить від присутності інших молекул та залежить від відстані між поверхнею та молекулою адсорбату.
 ,
,
 - Адсорбційний потенціал.
- Адсорбційний потенціал.
Оскільки поверхня неоднорідна, відстань замінюють на адсорбційний об'єм.  .Адсорбційний обсяг– це обсяг, укладений між поверхнею та точкою, що відповідає даному значенню
.Адсорбційний обсяг– це обсяг, укладений між поверхнею та точкою, що відповідає даному значенню  .
.
Адсорбційний потенціал– це робота перенесення 1 моль адсорбату поза даним адсорбційним об'ємом у цю точку адсорбційного об'єму (або робота перенесення 1 моль насиченої пари адсорбату, що знаходиться в рівновазі з рідким адсорбатом без адсорбенту в рівноважну з адсорбентом парову фазу).

Характеристична крива
 - адсорбційний потенціал,
- адсорбційний потенціал, 
Для даного адсорбенту та різних адсорбатів справедливо: 
Для різних типів адсорбатів  ,
,
де  потенціали для ізотерм адсорбції при відносних тисках
потенціали для ізотерм адсорбції при відносних тисках  для адсорбату 1 і адсорбату 2. Це ставлення є величина постійна.
для адсорбату 1 і адсорбату 2. Це ставлення є величина постійна.
 - Коефіцієнт афінності
- Коефіцієнт афінності
Теорія капілярної конденсації.
Перебіг процесу адсорбції багато в чому залежить від структури пористого тіла.
|
Мікропористі | |
|
Перехіднопористі | |
|
Макропористі |
У разі мікропористих сорбентів поля адсорбційних сил перекриваються. У разі макропористих сорбентів пори виконують роль транспортних каналів. Процеси коденсації найбільш значущі в перехіднопористих тілах. Капілярна конденсація починається за певних значень pі  , коли частина поверхневої енергії вже компенсована. Необхідна умова – поверхня має бути самчується. Процеє описується рівнянням Томпсона – Кельвіна.
, коли частина поверхневої енергії вже компенсована. Необхідна умова – поверхня має бути самчується. Процеє описується рівнянням Томпсона – Кельвіна.

 - для випадку змочування центр кривизни знаходиться в газовій фазі.
- для випадку змочування центр кривизни знаходиться в газовій фазі.
У разі капілярної конденсації ізотерму адсорбції має гістерезисний вигляд. Процес адсорбції відповідає нижня гілка, процесу десорбції - верхня.
Усі види пір можна звести до трьох видів:
|
Конічні |
Циліндричні з одним закритим кінцем |
Циліндричні з двома відкритими кінцями |
|
Процесне наповнення здійснюється з дна пори. Ізтерма адсорбції та ізотерма десорбції в цьому випадку збігаються, оскільки процес адсорбції починається зі сфери та процес десорбції також починається зі зникнення деяких сфер.
↓ |
Гістерези немає. Прямий та зворотний хід описуються рівнянням:
|
Дна ніде немає, заповнення пори піде стінками циліндра.
циліндр: Ізотерма і матиме гістерезисний вигляд.
↓ |


 - сфера,
- сфера, ,
,



У  умовах змочування конденсація здійснюється за нижчих тисків, що енергетично вигідно. По десорбційної гілки одержують криві розподілу пір за розмірами.
умовах змочування конденсація здійснюється за нижчих тисків, що енергетично вигідно. По десорбційної гілки одержують криві розподілу пір за розмірами.
Максимум диференціальної кривої зміщений вліво щодо точки інтегральної перегину. Загальний обсяг малих пір невеликий, проте має великі значення площі поверхні. Зі збільшенням розміру доби, їх обсяг зростає як  , а площа як
, а площа як  , за рахунок цього і спостерігається усунення максимуму диференціальної кривої.
, за рахунок цього і спостерігається усунення максимуму диференціальної кривої.
Адсорбція межі тверде тіло – рідина.
У разі адсорбції на кордоні тверде тіло – газ ми нехтували одним компонентом. У разі адсорбції на кордоні тверде тіло – рідина адсорбат витісняє з поверхні адсорбенту молекули розчинника.
 ,
,
Справедливе рівняння:

 ,
,
N 1 , N 2 – мольні частки розчинника та компонента, N 1 + N 2 = 1, тоді
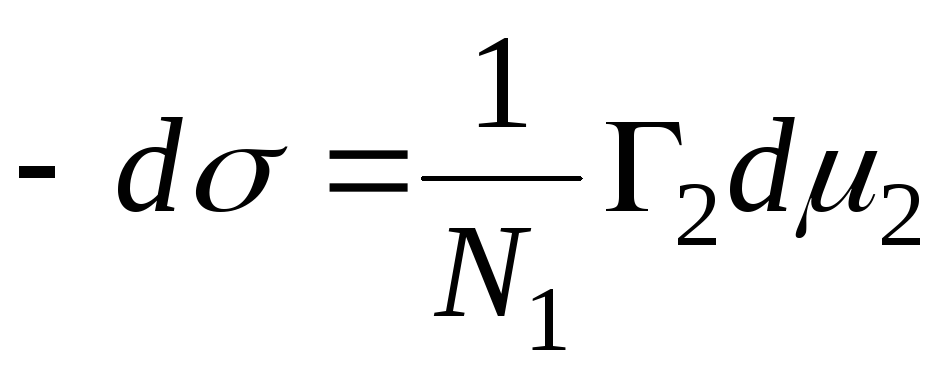 ,
=>
,
=>
 , Тоді-рівняння адсорбції для межі розділу фаз тверде тіло - рідина.
, Тоді-рівняння адсорбції для межі розділу фаз тверде тіло - рідина.
Адсорбція (Г) > 0 при  <
0
<
0
Якщо значення  для компонента та розчинника сильно різні, у цьому випадку залежність Гвід Nмає екстремум при значенні N
~ 0,5.
для компонента та розчинника сильно різні, у цьому випадку залежність Гвід Nмає екстремум при значенні N
~ 0,5.
Е  слі
слі  мають близькі значення, у разі можлива зміна знака адсорбції. Залежність Гвід Nперетинає вісь абсцис
мають близькі значення, у разі можлива зміна знака адсорбції. Залежність Гвід Nперетинає вісь абсцис
Точка перетину функції Г(N) з віссю абсцис називається адсорбційним азеотропом. Це означає, що два компоненти не можуть бути поділені на даному адсорбенті.
Рівняння ізотерми адсорбції із константою обміну.
При адсорбції на кордоні тверде тіло – рідина постійно відбувається перерозподіл компонентів між поверхнею адсорбенту та об'ємом розчину.
 - компоненти (- - відносяться до поверхні)
- компоненти (- - відносяться до поверхні)

 ,
,
 ,
, .
.
 ,
,


Адсорбція на кордоні рідина-газ
Р  Розглянемо зміну концентраційного профілю в міру перетину кордону розділу рідина-газ. Нехай компонент 2 лет.
Розглянемо зміну концентраційного профілю в міру перетину кордону розділу рідина-газ. Нехай компонент 2 лет.
Cs – концентрація у поверхневому шарі.
Виходячи з визначення надмірної адсорбції

Якщо компонент не леткий, то величина адсорбції запишеться так:
П  рі
рі 

У рівнянні  природа речовини описується похідною
природа речовини описується похідною  .
.
Ізотерма поверхневого натягу може мати вигляд 1 або 2:
1 – поверхнево-інактивні речовини
2 – поверхнево-активні речовини
Поверхневою активністю g називається здатність речовин знижувати поверхневий натяг у системі.

 - Товщина поверхневого шару
- Товщина поверхневого шару
C s- Концентрація компонента в поверхневому шарі
З– об'ємна концентрація
Для гомологічного ряду існує правило:
 - правило Траубо Дюкло
- правило Траубо Дюкло
Для гомологічного ряду ізотерму адсорбції має такий вигляд:
Замість A пишемо Р, оскільки адсорбція надлишкова у поверхневому шарі.
Ізотерма поверхневого натягу:

 - Поверхневий натяг чистого розчинника.
- Поверхневий натяг чистого розчинника.
 - фундаментальне адсорбційне рівняння;
- фундаментальне адсорбційне рівняння;
 - Рівняння Лангмюра.
- Рівняння Лангмюра.
Вирішимо їх спільно:

- рівняння Шишковського.
B- Константа для гомологічного ряду.
A- при переході від одного гомолога до іншого збільшується у 3-3,5 рази 
![]()
1 – область малих концентрацій
![]()
2 – середня концентрація
3 – мономолекулярний шар
Поверхневоактивні речовини є дифільні молекули, тобто. включають полярну групу та неполярний вуглеводневий радикал.
o – полярна частина молекули.
| - Неполярна частина молекули.
У полярному розчиннику молекули ПАР орієнтуються таким чином, що полярна частина молекули звернена до розчинника, а неполярна виштовхується в газову фазу.
У рівнянні Шишковського  вона постійна для гомологічного ряду.
вона постійна для гомологічного ряду.
Поверхнево-активна дія починає проявлятися з n>5. При концентраціях більших, ніж концентрація мономолекулярного шару, в розчинах ПАР відбувається міцелоутворення.
Міцелла- називається агрегат молекул дифільних ПАР, вуглеводневі радикали яких утворюють ядро, а полярні групи звернені у водну фазу.
Маса міцели – міцеляльна маса.
Ч  сло молекул – число агрегації.
сло молекул – число агрегації.
Сферичні міцели
У разі міцелоутворення в розчині встановлюється рівновага
ККМ – критична концентрація міцелоутворення.
Оскільки ми вважаємо міцелу окремою фазою:
Для гомологічного ряду існує емпіричне рівняння:
a- Енергія розчинення функціональної групи.
b – інкремент адсорбційного потенціалу, робота адсорбції на одну метиленову ланку.
– інкремент адсорбційного потенціалу, робота адсорбції на одну метиленову ланку.
Наявність у міцелах вуглеводневого ядра створює можливість для розчинення у водних розчинах ПАР сполук, які не розчиняються у воді, це явище називається солюбілізацією (те, що розчиняється – солюбілізат, ПАР – солюбілізатор).
Бруд може бути зовсім не полярний, може містити як полярну, так і не полярну частину і орієнтуватиметься як молекула ПАР.
У будь-якому випадку при солюбілізації відбувається збільшення міцелярної маси та числа агрегації не тільки за рахунок включення солюбілізату, але й за рахунок збільшення числа молекул ПАР, необхідних підтримки рівноважного стану.
Солюбілізація тим ефективніше, що менше молекулярна маса солюбілізату.

 ~ 72 мН\м.
~ 72 мН\м.
 ~ 33 мН\м.
~ 33 мН\м.
Ефективність ПАР залежить від величини ККМ.
Двовимірний тиск поверхневого шару

→ -Сили поверхневого натягу.
- двомірний тиск.
Поверхневий шар - це сила, що дорівнює різниці поверхневих натягів розчину ПАР і чистого розчинника, спрямована в бік чистої поверхні.
Між розчином та поверхневим шаром встановлюється рівновага



При  існує область, де
існує область, де  лінійно залежить від концентрації.
лінійно залежить від концентрації.
Р [моль/м 2 ].
 -Площа, займана одним молем речовини
-Площа, займана одним молем речовини
Тоді ізотерма двовимірного тиску матиме вигляд 
 - Ізотерма двомірного тиску.
- Ізотерма двомірного тиску.
Залежність  від S М:
від S М:

При  - Двовимірний тиск різко зростає. При
- Двовимірний тиск різко зростає. При  двовимірний деформується, що викликає різке зростання
двовимірний деформується, що викликає різке зростання  .
.
Плівка по обидва боки обмежена однаковими фазами називається двосторонньою. У таких плівках спостерігається постійний рух маткового розчину.
Плівки завтовшки менше 5 нм називаються чорними плівками.

Адсорбційні шари повинні мати дві характеристики: в'язкість і легкорухливість, плинність і пружність.
Ефект Марангоні – це самозалікування.
Трикутник Гіббса,  - надлишковий тиск.
- надлишковий тиск.
Плівка розтягнулася і за рахунок того, що частина рідини пішла, ПАР усуваються у вільне місце. Трикутник Гіббса.
Ефект адсорбційної міцності тел.
На поверхні плівки завжди існує адсорбційний шар, для якого тоді
Рівняння Лангмюра:


 у двомірний тиск
у двомірний тиск
 - аналог рівняння Шишковського
- аналог рівняння Шишковського
Електрокінетичні явища. Подвійний електричний прошарок (ДЕС).
Модель Гелемгольця. Теорія Гуї-Чапмена.
1808 р. Рейс

U – образна трубка, занурюють у неї 2 електроди. Закон сполучених судин порушується і відбувається зміна рівня рідини у трубці – електрокінетичні явища.
Кінетичні явища:
Електрофорез
Електроосмос
Потенціал течії (протікання)
Потенціал седиментації
1 і 2 виникають при накладенні різниці потенціалів, 3 і 4 продавлювання та седиментація колоїдних частинок викликають появу різниці потенціалів.
Електроосмосом називається рух дисперсійного середовища щодо нерухомої дисперсної фази під дією електричного струму.
Електрофорез – це рух частинок дисперсної фази щодо нерухомого дисперсійного середовища під дією електричного струму.
П  ричина виникнення електрокінетичних явищ – це просторовий поділ зарядів та виникнення подвійного електричного шару.
ричина виникнення електрокінетичних явищ – це просторовий поділ зарядів та виникнення подвійного електричного шару.
Подвійний електричний шар є плоским конденсатором, одна обкладка утворена потенціалорозділяючими іонами, інша - протиіноами. Іони заражені також як потенціалозначальні ко-іони відтіснені в об'єм розчину. Відстань між обкладками  . Потенціал падає лінійно, різниця потенціалів
. Потенціал падає лінійно, різниця потенціалів  .
.
Зовнішня різниця потенціалів викликає появу модуля зсуву  - це пара сил віднесених до одиниці площі, що діють уздовж поверхні твердого тіла.
- це пара сил віднесених до одиниці площі, що діють уздовж поверхні твердого тіла.

У стані рівноваги модуль зсуву дорівнює модулю в'язкого тертя (  ).
).

У наших умовах  ,
,

 - рівняння Гелемгольця-Смалуковського
- рівняння Гелемгольця-Смалуковського
 - Лінійна швидкість зміщення я фаз.
- Лінійна швидкість зміщення я фаз.
E- Напруженість електричного поля.
 - Різниця потенціалів між обкладками
- Різниця потенціалів між обкладками
 - електрофоретична рухливість [м 2 / (В * с)].
- електрофоретична рухливість [м 2 / (В * с)].
У моделі Гелемгольця не враховується тепловий рух молекул. Реально розподіл іонів у подвійному шарі має більш складний характер.
Гуї та Чапман виділили такі причини виникнення ДЕС:
Перехід іона з однієї фази до іншої при встановленні рівноваги.
Іонізація речовини твердої фази.
Добудовує поверхню іонами, присутніми в дисперсійному середовищі.
Поляризація від зовнішнього джерела струму.
Подвійний електричний шар має розмиту чи дифузну будову. Іони прагнуть поступово розподілитися у всьому дифузному шарі.
Дифузний шар складається з протиінов, протяжність шару визначається їх кінетичною енергією. При температурі протиіної, що прагне до абсолютного нуля, максимально наближені до потенціаловизначальних іонів.
Даня теорія базується на двох рівняннях:
рівняння Больцмана

 - Праця проти сил електростатичної взаємодії.
- Праця проти сил електростатичної взаємодії.
 - Об'ємна щільність заряду.
- Об'ємна щільність заряду.

Рівняння Пуассона 
Оскільки товщина ДЕС набагато менша за розміри частинки і для плоского ДЕС похідна за координатами  і
і  скасовується.
скасовується. 
Для е у при у<<1 функцию можно разложить в ряд Маклорена:

Обмежимося двома членами ряду, тоді:


 - Товщина ДЕС - це відстань, на якій потенціал ДЕС зменшується в eразів.
- Товщина ДЕС - це відстань, на якій потенціал ДЕС зменшується в eразів.
Чим менша температура, тим менше  . При Т→0 – плаский ДЕС. Чим більша концентрація, тим більше I, тим менше
. При Т→0 – плаский ДЕС. Чим більша концентрація, тим більше I, тим менше  .
.





 "-" означає, що потенціал з відстанню зменшується. =>
"-" означає, що потенціал з відстанню зменшується. =>
=>

 ,
,
 - Потенціал експоненційно зменшується.
- Потенціал експоненційно зменшується.
Потенціал для поверхневої щільності заряду: 
Поверхневий заряд – об'ємний заряд із протилежним знаком, проінтегрований на відстані.




=>


Там, де потенціал зменшується у 2,7 раза - 
Ємність подвійного шару 
Недолік теорії – не враховується наявність прошарку Гелемгольца, тобто. не враховує  звідси помилки у визначенні основних параметрів. Також пояснює вплив іонів різної природи на товщину подвійного електричного шару.
звідси помилки у визначенні основних параметрів. Також пояснює вплив іонів різної природи на товщину подвійного електричного шару.
Теорія Штерна. Будова колоїдної міцели.
Подвійний електричний шар складається з двох частин: щільної та дифузної. Щільний шар утворюється в результаті взаємодії потенціалутворюючих іонів зі специфічно адсорбуються. Ці іони, як правило, частково або повністю дегідратовані і можуть мати як однаковий, так і протилежний до іонів потенційно заряд. Це залежить від співвідношення енергії електростатичної взаємодії  та потенціалу специфічної адсорбції
та потенціалу специфічної адсорбції  . Іони щільного шару закріплені. Інша частина іонів розташована в дифузному шарі, ці іони є вільними і можуть переміщатися вглиб розчину, тобто. з області більшої концентрації в область меншої. Загальна щільність заряду складається із двох частин.
. Іони щільного шару закріплені. Інша частина іонів розташована в дифузному шарі, ці іони є вільними і можуть переміщатися вглиб розчину, тобто. з області більшої концентрації в область меншої. Загальна щільність заряду складається із двох частин.

 -заряд шару Гельмгольця
-заряд шару Гельмгольця
 -Заряд дифузного шару
-Заряд дифузного шару
Поверхня має кілька адсорбційних центрів, кожен із яких взаємодіє з одним протиіоном. Константа такої квазіхімічної реакції дорівнює:

 , де
, де  - мольна частка протиіонів у розчині
- мольна частка протиіонів у розчині
Розподіл Гельмгольця
Потенціал зменшується лінійно
Розподіл потенціалу по Гуї. Щільного шару немає, потенціал зменшується експоненційно зі значення 
Розподіл по Штерну.
Спочатку зниження потенціалу лінійне, та був експоненційно.
При накладенні електричного поля у разі електрофорезу рухається не безпосередньо частка твердої фази, а частка твердої фази з шаром іонів, що її оточують. ДЕС повторює форму частинки дисперсної фази. При накладенні потенціалу відривається частина дифузного шару. Лінія розриву називається кордоном ковзання.
Потенціал, що виникає на межі ковзання внаслідок відриву частини дифузного шару, називається електрокінетичним потенціалом(Дзета потенціал  ).
).
Частка дисперсної фази, з навколишнім шаром протиіонів і подвійним електричним шаром називається міцелою.
Правила написання колоїдних міцел:


1-1 зарядний електроліт
T – частка дисперсної фази.
AA – межа щільної та дифузної частини.
BB – межа ковзання.
Кордон ковзання може збігатися з лінією AA, а може й не збігатися.
Значення pH, при якому дзета-потенціал дорівнює нулю, називається ізоелектричною точкою.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. У надлишку CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m·nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - Запис міцели.
CaSO 4 m – агрегат.
CaSO 4 m∙nCa 2+ – ядро.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - частка.
2. У надлишку Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - міцелу
CaSO 4 m – агрегат.
CaSO 4 m∙nSO 4 2 + – ядро.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - частка
Рівняння Гелемгольця-Смолуховського

 - Лінійна швидкість зміщення кордонів (в електроосмосі).
- Лінійна швидкість зміщення кордонів (в електроосмосі).
 - Різниця потенціалів на обкладках конденсатора (в електроосмосі).
- Різниця потенціалів на обкладках конденсатора (в електроосмосі).


 - об'ємна швидкість перебігу розчину, S- Площа поперечного перерізу комірки.
- об'ємна швидкість перебігу розчину, S- Площа поперечного перерізу комірки.

E- Напруженість електричного поля.


 (Для електроосмосу).
(Для електроосмосу).
Для потенціалу течії:

 - потенціал
- потенціал
 - тиск на мембрану
- тиск на мембрану
Як правило, значення електрофоретичних рухливостей та електроосмотичних рухомостей менше розрахункових. Це відбувається внаслідок:
Релаксаційного ефекту (під час руху частки дисперсної фази порушується симетрія іонної атмосфери).
Електрофоретичного гальмування (виникнення додаткового тертя внаслідок руху протиіонів).
Спотворення ліній струму у разі електропровідних частинок.
Зв'язок поверхневого натягу з потенціалом. Рівняння Ліппмана.
Освіта ДЕС відбувається спонтанно внаслідок прагнення системи знизити свою поверхневу енергію. В умовах сталості Tі pузагальнене рівняння першого та другого законів термодинаміки виглядає:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1-е рівняння Ліппмана.
- 1-е рівняння Ліппмана.
 - Поверхнева щільність заряду.
- Поверхнева щільність заряду.
 - Диференційна ємність.
- Диференційна ємність.
 - 2-е рівняння Ліппмана.
- 2-е рівняння Ліппмана.
З- Місткість.
Вирішимо 1-е рівняння Ліппмана та фундаментальне рівняння адсорбції:
 ,
,


 тоді
тоді
 - Рівняння Нернста
- Рівняння Нернста
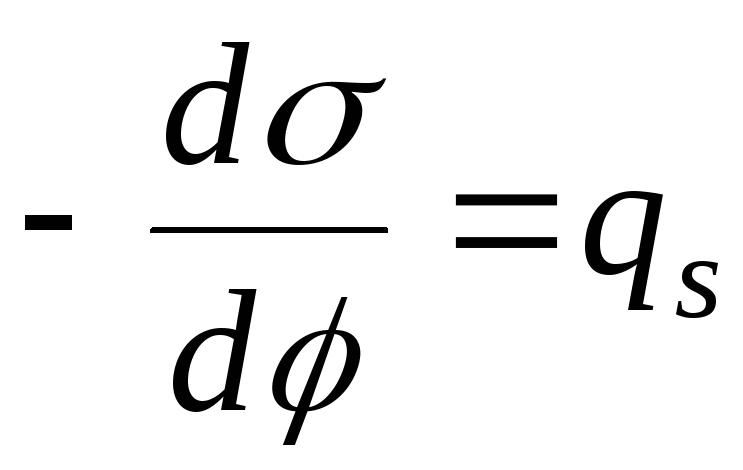 ,
,
 ,
,




 - Рівняння електрокапілярної кривої (ЕКК).
- Рівняння електрокапілярної кривої (ЕКК).
У  :
: , але
, але 
Катіонні ПАР (КПАВ) знижують катодну гілку ЕКК.
Аніонні ПАР (АПАВ) знижують анодну гілку ЕКК.
Неіоногенні ПАР (НПАВ) знижують середню частину ЕКК.
Стійкість дисперсних систем. Тиск, що розклинює.
Дисперсні системи можна поділити:
Системи нестійкі термодинамічно можуть бути кінетично стійкі за рахунок переходу в метастабільний стан.
Розрізняють два види стійкості:
Седиментаційна стійкість (стосовно силі тяжкості).
Агрегативна стійкість. (стосовно злипання)
Коагуляція- Це процес злипання частинок, що призводить до втрати агрегатної стійкості. Коагуляцію може спричинити зміну температури, pH, перемішування, ультразвук.
Розрізняють коагуляцію:
Оборотна.
Необоротна.
Коагуляція протікає під час введення електролітів.
Правила коагуляції:

Плівка- Це частина системи, що знаходиться між двома міжфазними поверхнями.
тиск, що розклинюєвиникає при різкому зменшенні товщини плівки в результаті взаємодії поверхневих шарів, що зближуються.

«-» - при зменшенні товщини плівки тиск, що розклинює, зростає.
P 0 - Тиск в об'ємній фазі, яка є продовженням прошарку.
P 1 – тиск у плівці.
Теорія стійкості. ДЛФО (Дерягін, Ландау, Фервей, Овербек).
Відповідно до теорії ДЛФО у розклинювальному тиску виділяють дві складові:
ЕлектростатичнаП Е (позитивна, вона обумовлена силами електростатичного відштовхування). Відповідає зменшенню енергії Гіббса у разі зростання товщини плівки.
МолекулярнаП М (негативна, обумовлена дією сил тяжіння). Обумовлена стиском плівки за рахунок хімічних поверхневих сил, радіус дії сил десяті долі нм з енергією близько 400 кДж/моль.
Повна енергія взаємодії:

 - система агрегативно стійка
- система агрегативно стійка
 - нестійка система
- нестійка система
П  оложна складова.
оложна складова.
Збільшення обумовлено збільшенням потенційної енергії при стисканні тонких плівок. Для плівок великої товщини надлишкова енергія іонів скомпенсована і дорівнює енергетичній взаємодії в об'ємі дисперсійного середовища.
Якщо  (
( - Товщина плівки,
- Товщина плівки,  - радіус іона) утончення плівки призводить до зникнення та зменшення в ній молекул та іонів з мінімальною поверхневою енергією. Число сусідніх частинок зменшується, внаслідок чого потенційна енергія частинок, що залишилися в плівці, зростає.
- радіус іона) утончення плівки призводить до зникнення та зменшення в ній молекул та іонів з мінімальною поверхневою енергією. Число сусідніх частинок зменшується, внаслідок чого потенційна енергія частинок, що залишилися в плівці, зростає.


Теорія ДЛФО взаємодія частинок розглядає як взаємодію пластин.

Частинки не взаємодіють



 - Рівняння Лапласа,
- Рівняння Лапласа,  ,
,


Для слабко заряджених поверхонь
Для сильно заряджених поверхонь:

Молекулярна складова – взаємодія двох атомів:
 ~
~
Взаємодія атома з поверхнею:

Візьмемо дві платівки:
Д  ля отримання молекулярної складової необхідно провести підсумовування всіх енергій взаємодії атомів правої та лівої пластин.
ля отримання молекулярної складової необхідно провести підсумовування всіх енергій взаємодії атомів правої та лівої пластин.
де  - Постійна Гамакера (враховує природу тіл, що взаємодіють).
- Постійна Гамакера (враховує природу тіл, що взаємодіють).
Т.о. Енергія взаємодії частинок у системі може бути виражена за допомогою потенційних кривих.
I – первинний потенційний мінімум. Це зона незворотної коагуляції, сили тяжіння переважають.
II – зона агрегативної стійкості, що переважають сили відштовхування.
ІІІ – вторинний потенційний мінімум (або зона флокуляції). Між частинками дисперсної фази існує прошарок електроліту, і частинки можуть бути розділені та переведені в зону агрегативної стійкості.

Крива 1 – система агрегативно стійка.
Крива 2 – у зоні I стійка, у зоні II не стійка.
Крива 3 – у системі відбулася коагуляція.
Крива 4 – у точці 4 сумарна енергія взаємодії U = 0,  ця точка екстремуму відповідає початку швидкої коагуляції.
ця точка екстремуму відповідає початку швидкої коагуляції.
Існує два випадки:
1. Поверхні слабозаряджені:
U = U Е + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - це товщина прошарку, що відповідає початку процесу коагуляції.
- це товщина прошарку, що відповідає початку процесу коагуляції.






 - для слабозаряджених поверхонь
- для слабозаряджених поверхонь
 тоді
тоді 
2. Для сильнозаряджених поверхонь:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Зведемо (3) у квадрат

Коагуляція:
При специфічній адсорбції іони можуть адсорбуватись у надеквівалентній кількості таким чином, що поверхня може змінити свій заряд. Відбувається перезаряджання поверхні.
У разі специфічної адсорбції можуть адсорбуватися іони як протилежних знаків, а й одного.
Якщо адсорбуються іони того ж знака, що й поверхня, то поверхневому шарі відбуватиметься не падіння потенціалу, а його зростання.

Нейтралізаційна коагуляція (протікає за участю слабозаряджених частинок і залежить не тільки від заряду електроліту-коагулятора, а й від потенціалу на межі щільного та дифузного шару).
Теорія швидкої коагуляції Смолуховського.

Залежність швидкості коагуляції від концентрації електроліту.
I – швидкість коагуляції мала,
II – швидкість коагуляції практично пропорційна концентрації електроліту.
III – область швидкої коагуляції, швидкість практично залежить від концентрації.
Основні положення:
Вихідний монодисперсний золь, подібні частинки мають сферичну форму.
Усі зіткнення часток результативні.
При зіткненні двох первинних частинок утворюється вторинна. Вторинна + первинна = третинна. Первинне, вторинне, третинне – кратність.
У термінах хімічної кінетики процес коагуляції може бути описаний рівнянням:
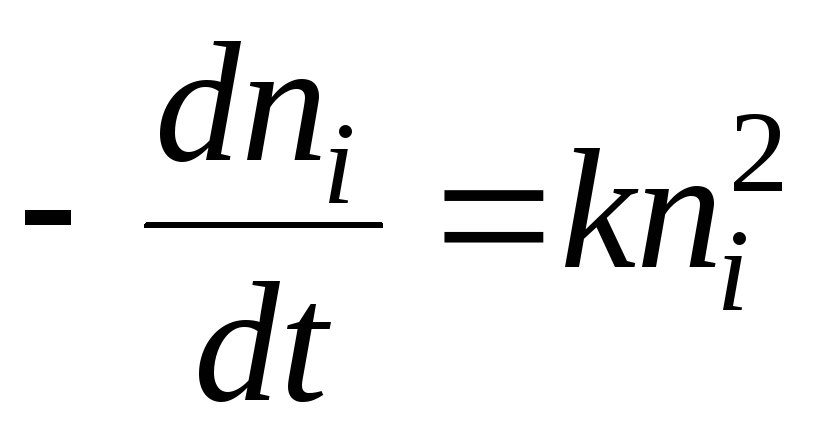
Рішенням буде рівняння: 
 - Час половинної коагуляції. Це час, протягом якого кількість частинок золю зменшується в 2 рази.
- Час половинної коагуляції. Це час, протягом якого кількість частинок золю зменшується в 2 рази.

 ,
,
 ,
,
 ,
,


У міру збільшення кратності максимум кривих коагуляції зрушується у бік більших значень  .
.
Недоліки:
Припущення про монодисперсність.
Припущення результативності всіх зіткнень.


