Adsorbimi bëhet në ndërfaqe. Prandaj, është e arsyeshme të konsiderohet përshkrimi termodinamik i dukurive sipërfaqësore si një rast i veçantë i termodinamikës së sistemeve heterogjene.
Oriz. 3.4. Adsorbimi Gibbs: 1- sistem krahasimi dyfazor, 2- sistem i vërtetë dyfazor me një rajon jo uniform
Në termodinamikën e sistemeve heterogjene përdoret parimi i aditivitetit e cila është si më poshtë: të gjitha vetitë ekstensive të një sistemi heterogjen janë të barabarta me shumën e vetive ekstensive përkatëse që do të kishin pasur fazat përpara se të viheshin në kontakt. Le t'i shënojmë fazat me α dhe β (Fig. 4). Pastaj për një sistem ideal, i tillë që vetitë e fazave pranë ndërfaqes të përkojnë me vetitë e tyre në masë, relacionet e mëposhtme janë të vlefshme për energjinë e brendshme U, vëllimin V, masën (numrin e moleve) n, entropinë S pas vendosjes së ekuilibrit në një sistem heterogjen:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Kjo supozon se temperatura dhe presioni në të dy fazat janë të njëjta.
Për sistemet reale heterogjene, rajoni i tranzicionit në kufirin e dy fazave jep një kontribut shtesë në vetitë e gjera të sistemit. Nëse ndodhin fenomene sipërfaqësore, duhet të merret parasysh ndryshimi midis vetive të gjera të një sistemi real heterogjen dhe vetive të gjera të një sistemi model në të cilin dukuritë sipërfaqësore mungojnë. Një sistem i tillë quhet sistem krahasimi. Sistemi i krahasimit ka të njëjtat parametra intensivë (T, P, C i ...) dhe të njëjtin vëllim V si sistemi real (Fig. 4).
Nga pikëpamja termodinamike, vlera e përthithjes G kuptohet si sasia e tepërt e substancës n s, e shprehur në mole ose gram, që ka një sistem real heterogjen në krahasim me sistemin e referencës, në lidhje me zonën e ndërfaqes ose me sipërfaqen. i adsorbentit A. Supozohet se sistemi i krahasimit ka të njëjtat parametra intensivë (T, P, C i), dhe të njëjtin vëllim (V = V α + V β) si sistemi real (Fig. 4) .
Г = (n - n α - n β)/A = n s /A 3.11
Funksionet termodinamike të tepërta të rajonit të tranzicionit të një sistemi real (i shënojmë me indeksin s) mund të shkruhen si
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β etj.
Matjet eksperimentale të adsorbimit japin gjithmonë adsorbimin saktësisht si një tepricë e komponentit në sistemin real në krahasim me sistemin e zgjedhur të referencës. Për shembull, kur adsorbimi i gazit në një adsorbent të ngurtë ose kur adsorbimi i përbërësve në një fazë të ngurtë, për të gjetur vlerat e absorbimit, përcaktohet ndryshimi në përqendrimet fillestare të adsorbatit pas kontaktit të fazave α dhe β.
n i s = V(C i o - C i),
Ku C i o– përqendrimi fillestar i komponentit të i-të, C i– përqendrimi i komponentit të i-të pas vendosjes së ekuilibrit ndërmjet fazave të kontaktit. Besohet se vëllimi V nuk ndryshon. Megjithatë, përqendrimi i komponenti i th C i, i marrë në mënyrë eksperimentale, përcaktohet në vëllim V' mbi ndërfaqen e fazës pa marrë parasysh vëllimin e rajonit johomogjen të shtresës së tranzicionit V α në ndërfaqen ku është përqendrimi C i α. Kështu, për shkak të ekzistencës së një rajoni jo uniform në një sistem real, vëllimi i përgjithshëm i sistemit mund të përfaqësohet si V = V’ + V α. E gjithë sasia i-komponenti C i o do të shpërndahet ndërmjet këtyre dy vëllimeve:
V C i o = V’ C i + V α C i α,
dhe numri i moleve të komponentit i, i adsorbuar në ndërfaqe, do të jetë i barabartë me
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
ato. Adsorbimi i përcaktuar në mënyrë eksperimentale është teprica e komponentit të i-të në vëllimin V α në krahasim me sasinë e këtij komponenti në të njëjtin vëllim larg ndërfaqes së fazës. Ky lloj adsorbimi quhet adsorbimi Gibbs. .
V α C i α quhet përmbajtje e plotë i- Komponenti i th ne shtresen e adsorbimit. Në rajonin e përqendrimeve shumë të ulëta C i në vëllim V' amendament V α C i ekuacioni (3.2) mund të neglizhohet dhe vlera e matur mund të merret parasysh V α C i α përmbajtje të plotë i- Komponenti i th në shtresën e adsorbimit, për shembull, gjatë thithjes së gazit në një adsorbent të ngurtë në presione të ulëta.
Termodinamika e proceseve të përthithjes.
| Emri i parametrit | Kuptimi |
| Tema e artikullit: | Termodinamika e proceseve të përthithjes. |
| Rubrika (kategoria tematike) | Arsimi |
Përkufizimet bazë dhe metodat e klasifikimit të proceseve të adsorbimit.
Adsorbimi i referohet fenomeneve që ndodhin për shkak të një rënie spontane të energjisë sipërfaqësore.
Adsorbimi– procesi i rishpërndarjes spontane të kthyeshme ose të pakthyeshme të përbërësve të një sistemi heterogjen ndërmjet shtresës sipërfaqësore dhe vëllimit të fazës homogjene.
Në sistemet me shumë komponentë, komponenti që redukton më fuqishëm tensionin ndërfaqësor preferohet të transferohet në shtresën sipërfaqësore. Në sistemet me një përbërës, gjatë formimit të shtresës sipërfaqësore, ndodh një ndryshim në strukturën e saj (një orientim i caktuar i atomeve dhe molekulave, polarizimi), i quajtur autoadsorbimi.
Faza më e dendur në të cilën lokalizohen ndërveprimet e adsorbimit quhet adsorbent. Substanca e rishpërndarë midis vëllimit të fazës homogjene dhe shtresës sipërfaqësore përcaktohet me termin ʼʼ adsorbojʼʼ.
Në disa raste, procesi i përthithjes është i kthyeshëm. Në këtë rast, në kushte të caktuara, një pjesë e molekulave të përthithura si rezultat i fenomeneve kinetike molekulare mund të lëvizin nga shtresa sipërfaqësore në fazën më të madhe. Procesi i kundërt i adsorbimit quhet desorbimi.
Metodat për klasifikimin e proceseve të adsorbimit.
Klasifikimi i proceseve të adsorbimit sipas gjendjes së grumbullimit të fazave ndërvepruese. Duke marrë parasysh varësinë nga gjendja agregate e fazave ngjitur, dallohen llojet e mëposhtme të proceseve të adsorbimit:
Adsorbimi i gazrave në adsorbentë të ngurtë;
Adsorbimi i substancave të tretura në ndërfaqet "ngurtë-lëng" dhe "lëng-lëng";
Adsorbimi i surfaktantëve në ndërfaqen e lëngët-gazit.
Klasifikimi i proceseve të adsorbimit sipas mekanizmit të ndërveprimit ndërmjet adsorbentit dhe adsorbatit. Adsorbimi mund të konsiderohet si ndërveprim i molekulave të adsorbatit me qendrat aktive të adsorbentit. Sipas mekanizmit të ndërveprimit të tyre, ndahen llojet e mëposhtme të adsorbimit:
1) adsorbimi fizik (molekular).– ndërveprimi ndërmjet molekulave të adsorbatit dhe adsorbentit kryhet për shkak të forcave van der Waals, lidhjeve hidrogjenore (pa reaksione kimike);
2) adsorbimi kimik (kimisorbimi)- ngjitja e molekulave të adsorbatit në qendrat aktive të adsorbentit ndodh si rezultat i reaksioneve kimike të llojeve të ndryshme (me përjashtim të reaksioneve të shkëmbimit të joneve);
3) adsorbimi i shkëmbimit të joneve (shkëmbimi i joneve) - rishpërndarja e substancës adsorbatuese midis tretësirës dhe fazës së ngurtë (shkëmbyesi i joneve) sipas mekanizmit të reaksioneve të shkëmbimit të joneve.
Për të përshkruar në mënyrë sasiore proceset e adsorbimit, përdoren dy sasi.
1) Adsorbimi absolut– sasia (mol) ose masa (kg) e adsorbatit për njësi sipërfaqe ose masë të absorbuesit. Emërtimi – A; dimensioni: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Gibbs (tepricë) adsorbimi– teprica e substancës adsorbatuese në një shtresë sipërfaqësore me një trashësi të caktuar në krahasim me sasinë e saj në vëllimin e fazës homogjene, për njësi sipërfaqe ose masë të adsorbentit. Emërtimi – G; dimensioni: mol/m 2, mol/kᴦ.
Marrëdhënia midis adsorbimit absolut dhe të tepërt mund të ilustrohet duke përdorur ekuacionin:
Г = А – с * h (3.1)
ku c është përqendrimi ekuilibër i substancës në vëllimin e fazës, mol/m3;
h është trashësia e shtresës sipërfaqësore, e supozuar në mënyrë konvencionale të jetë 10 -9 m.
Në sistemet heterogjene shumëkomponente, kur një ose një përbërës tjetër rishpërndahet midis vëllimit të fazës homogjene dhe shtresës sipërfaqësore, ekuacioni për energjinë e tepërt të brendshme të sipërfaqes është i vlefshëm:
U = T * S + s * s + Sm i * n i (3.2)
Duke reduktuar të gjitha termat e ekuacionit në sipërfaqen njësi të sipërfaqes ndërfazore, marrim:
U s = T * S s + s + Sm i * Г i (3.3)
ku Г i = n i / s është teprica e komponentit i-të në shtresën sipërfaqësore, pra adsorbimi i Gibbs-it.
Për një sistem me një komponent, ekuacioni (3.3) do të marrë formën:
G s = s + m * G (3.4)
ku G s = U s - T * S s – Energjia Gibbs e sipërfaqes ose puna e krijimit të sipërfaqes së njësisë;
m * G – ngjeshja e substancës së substancës së përthithur në shtresën sipërfaqësore.
Bazuar në ekuacionin (3.4), mund të konkludojmë se gjatë adsorbimit, puna e krijimit të një sipërfaqeje ndërfazore konsiston në punën e formimit të sipërfaqes (prishja e lidhjeve kohezive në vëllimin e fazës së adsorbatit) dhe ngjeshja e substancës në shtresën sipërfaqësore.
Në një gjendje ekuilibri dinamik midis adsorbentit dhe adsorbatit, ndryshimi në energjinë Gibbs të sistemit heterogjen ΔG = 0, termodinamika e procesit të adsorbimit përshkruhet nga ekuacioni i quajtur Ekuacioni themelor i adsorbimit të Gibbs:
Ds = SГ i * dm i (3.5)
Ky ekuacion është universal, pasi është i vlefshëm për të gjitha llojet e proceseve të adsorbimit
Raste të veçanta të ekuacionit të adsorbimit Gibbs.
1) Adsorbimi nga solucionet.
Për potencialin kimik të komponentit të 1-të të sistemit gjatë adsorbimit në ndërfaqet "lëng-adsorbent i ngurtë" dhe "lëng-gaz", janë të vlefshme ekuacionet e mëposhtme:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
ku m i 0 është potenciali kimik i komponentit të i-të të sistemit në kushte standarde;
a i është aktiviteti i komponentit i-të të sistemit në kushte standarde.
Bazuar në këtë, ekuacioni i adsorbimit Gibbs merr formën:
Г i = - a i / R*T * (ds / da i) (3.8)
Për tretësirat e jo-elektroliteve marrim i = c i, atëherë:
Г i = - с / R*T * (ds / dс) (3.9)
Për tretësirat e elektrolitit:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
ku с ± është përqendrimi mesatar jonik i tretësirës;
n është koeficienti stekiometrik.
2) Adsorbimi i substancave nga faza e gazit.
Në përputhje me ekuacionin Mendeleev-Clayperon:
Р = с * R*T (3.11)
Në lidhje me këtë, ekuacioni Gibbs për përthithjen e gazeve në adsorbentët e ngurtë është shkruar në formën e mëposhtme:
Г i = - Р / R*T * (ds / dР) (3.12)
Në praktikë, ekuacioni i adsorbimit Gibbs lejon, bazuar në matjet e tensionit sipërfaqësor në vlera të ndryshme të përqendrimit të lëngut ose presionit të gazit të ekuilibrit, të llogaritet sasia e përthithjes së substancave në shtresën ndërfaqesore për të cilën përcaktohet tensioni sipërfaqësor.
Termodinamika e proceseve të përthithjes. - koncepti dhe llojet. Klasifikimi dhe veçoritë e kategorisë "Termodinamika e proceseve të adsorbimit". 2017, 2018.
Adsorbimi si përqendrim spontan i molekulave në një sipërfaqe shoqërohet me një ulje të entropisë së sistemit. Meqenëse kriteri për spontanitetin e procesit është
∆H - T · ∆S = ∆G< 0,
atëherë adsorbimi është i mundur vetëm në ∆H< 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. Me rritjen e temperaturës, ekuilibri zhvendoset drejt procesit endotermik, pra desorbimit.
Adsorbimi në një sipërfaqe të fortë
1. Adsorbimi monomolekular.
Sipas teorisë së Langmuir, molekulat adsorbente ndërveprojnë me sipërfaqen e adsorbentit, duke formuar përfundimisht një shtresë monomolekulare. Në këtë rast, shkalla e mbushjes () e sipërfaqes me substancën e absorbuar gjatë adsorbimit nga faza e gazit
nga lëngu
ku K është konstanta e ekuilibrit (konstanta e adsorbimit);
p është presioni i pjesshëm i gazit të përthithur;
c është përqendrimi i substancës së përthithur.
Varësia e β nga p (ose c) është paraqitur nga grafiku (izotermi i adsorbimit, T = konst) në Fig. 1.3.

Oriz. 1.3. Shkalla e mbushjes së sipërfaqes me substancë të përthithur
Në përqendrime të ulëta dhe presione të pjesshme, adsorbimi është proporcional me përqendrimin ose presionin e pjesshëm:
R<< 1, β ≈ К· r ilis<< 1, β ≈ К· s, d.m.th. seksioni fillestar i izotermës është afërsisht linear, dhe tan α = K (tg α përcaktohet nga pjerrësia e kurbës në p (ose c) → 0: ose ).
Nëse është numri i moleve të substancës së absorbuar për 1 g adsorbent; - numri maksimal i mundshëm i moleve të substancës së absorbuar për 1 g adsorbent (“kapaciteti i njështresor”), më pas
Zëvendësimi i β në ekuacionin (1.3) (për rastin e adsorbimit nga faza e gazit, përqendrimi Me në ekuacione duhet të zëvendësohet me presion R), marrim:
 (1.6)
(1.6)
Meqenëse dhe K në një çift të caktuar adsorbent-adsorbent janë konstante (at T=const), atëherë sipas varësisë mund të gjendet TE(Fig. 1.4).

Oriz. 1.4. Zgjidhja grafike e ekuacionit të adsorbimit
përftohet duke ekstrapoluar varësinë lineare eksperimentale në () = 0; dhe, që nga , atëherë , .
Vlera mund të përdoret për të përcaktuar sipërfaqen specifike të adsorbentit UD (në m 2 për 1 g adsorbent), nëse zona ω e zënë në sipërfaqe nga një molekulë e adsorbentit është e njohur (e përcaktuar nga madhësia e molekulës):
UD = · ω · Na, (1.7)
ku Na është numri i Avogadro-s (Na = 6.02 10 23).
Nga ana tjetër, vlera e njohur e UD mund të përdoret për të llogaritur ω të çdo substance bazuar në adsorbimin e saj në një adsorbent të caktuar.
2. Adsorbimi polimolekular.
Ekuacioni (1.5) përshkruan një kurbë me ngopje, d.m.th. në
p (ose c) → ∞ tenton në vlerën kufitare të barabartë me (Fig. 1.5,a).

Fig.1.5. Izotermat e adsorbimit:
a – adsorbimi me ngopje; b – adsorbimi polimolekular
Megjithatë, në disa raste, izotermat e adsorbimit duken si ato të paraqitura në Fig. 1.5, b, d.m.th. nuk e arrin kufirin edhe në p (ose c) të lartë.
Varësitë e tipit të paraqitur në Fig. 1.5,b korrespondojnë me adsorbimin polimolekular. Si rregull, izoterma të tilla janë karakteristike për substancat me ndërveprime të forta ndërmolekulare (për shembull, uji). Kur qendrat e adsorbimit në sipërfaqen e adsorbentit janë të zëna (shtresa monomolekulare është e ngopur), "ulja" e molekulave të ardhshme të adsorbatit ndodh për shkak të ndërveprimeve ndërmolekulare me molekulat tashmë të përthithura (Fig. 1.6). Nxehtësia e një adsorbimi të tillë është afër në vlerë absolute, por në shenjë e kundërt me nxehtësinë e avullimit të lëngut përkatës (mendoni pse).

Fig.1.6. Skema e adsorbimit:
a - adsorbimi monomolekular; b - adsorbimi polimolekular
Ndërsa afrohemi R ndaj presionit të avullit të ngopur të substancës së absorbuar, ajo fillon të kondensohet në sipërfaqen e adsorbentit, si rezultat, rritet me shpejtësi me rritjen R.
Në rastin e bashkëveprimit ndërmjet dy atomeve:
U – energjia e ndërveprimit;
U = U PARA. + U KTHIMI
 - Ekuacioni Lennard-Jones
, c, b, m = konst
- Ekuacioni Lennard-Jones
, c, b, m = konst
Në rastet e bashkëveprimit të atomeve me një sipërfaqe të fortë, është e nevojshme të përmblidhen të gjitha ndërveprimet.
x – distanca nga sipërfaqja
r – rrezja e veprimit të forcave tërheqëse
dV - vëllimi
n – numri i molekulave sipërfaqësore
U ADS. – energjia e ndërveprimit të adsorbimit



Në rastin e adsorbimit, tërheqja rritet. Dhe në rastin e ndërveprimit jopolar-jopolar, adsorbimi lokalizohet kryesisht në gropa.
Ndërveprimi elektrostatik.
Adsorbent polar - adsorbues jo polar
Adsorbues jo polar - adsorbues polar
Adsorbent polar - adsorbat polar.
M  Molekula e adsorbatit përfaqësohet si një dipol, dhe adsorbenti përfaqësohet si një përcjellës në të cilin molekula e adsorbatit indukton një pasqyrë dipole në mënyrë simetrike në lidhje me atë të dhënë.
Molekula e adsorbatit përfaqësohet si një dipol, dhe adsorbenti përfaqësohet si një përcjellës në të cilin molekula e adsorbatit indukton një pasqyrë dipole në mënyrë simetrike në lidhje me atë të dhënë.
X - distanca në mes
Kur ndërveprojmë, lind potenciali:
 ,
,
 - momenti dipol.
- momenti dipol.
Potenciali tenton të marrë vlerën maksimale, d.m.th. dipolet priren të orientohen pingul me sipërfaqen.
Meqenëse rritja e temperaturës nxit rritjen e lëvizjes Brownian, ajo çon në frenimin e procesit të adsorbimit.
Në rastin e ndërveprimit elektrostatik, adsorbati lokalizohet kryesisht në zgjatimet.
Ekuacioni themelor i adsorbimit.
Në rastin e adsorbimit, ndodh një rishpërndarje e komponentit, që do të thotë se potenciali kimik ndryshon. Procesi i adsorbimit mund të konsiderohet si kalimi i energjisë sipërfaqësore në energji kimike.
Vëllimi i shtresës = 0, pastaj ekuacioni i përgjithësuar i ligjeve I dhe II të termodinamikës:
T = konst; (1) = (2) => 

Për një sistem me dy komponentë:
,
 ,
,
 =>
=>
=>
 - Ekuacioni i adsorbimit të Gibbs
.
- Ekuacioni i adsorbimit të Gibbs
.
Për rastin e adsorbimit të TV. trup - gaz: , 
 ,
,

 - izotermi
- izotermi
 - izobar
- izobar
 - izopiknale
- izopiknale
 - isostere
- isostere
Izotermi, izopikni, izosteri janë të lidhura me njëra-tjetrën.
Sepse funksioni i adsorbimit 



Izotermia e Henrit Izotermia e Langmuirit



Termodinamika. Adsorbimi.
Për lëndën e kondensuar:
 ,
,
 ,
,
 - ndryshim integral në energjinë e Gibbs
.
- ndryshim integral në energjinë e Gibbs
.

P – presion mbi një sipërfaqe të lakuar, Р S – presion mbi një sipërfaqe të sheshtë
 - potenciali i përthithjes
- potenciali i përthithjes
Ndryshimi diferencial në kurth 
 , Г = konst
, Г = konst
- ndryshimi i entropisë diferenciale
- entalpi diferenciale e adsorbimit
 - nxehtësia izosterike e përthithjes
- nxehtësia izosterike e përthithjes
 - nxehtësia e kondensimit
- nxehtësia e kondensimit
 - nxehtësia neto e përthithjes
- nxehtësia neto e përthithjes

 ,
,




Qa - nxehtësia integrale e adsorbimit, 
Qra – nxehtësia neto integrale e adsorbimit,

ekuacioni i Henrit
Studimi i adsorbimit është i ndërlikuar nga heterogjeniteti i sipërfaqes, kështu që ligjet më të thjeshta fitohen për sipërfaqet homogjene.
Le të shqyrtojmë bashkëveprimin e gazeve me një sipërfaqe të ngurtë, kur një gaz kalon nga një gjendje ekuilibri në vëllim në një gjendje ekuilibri në sipërfaqe. Ky rast është analog me ekuilibrin e gazeve në një fushë graviteti.
 ,
,
 ,
=>
,
=> -ekuacioni i Henrit
-ekuacioni i Henrit
 - koeficienti i shpërndarjes
- koeficienti i shpërndarjes
Gjatë procesit të adsorbimit, ndodh një ndryshim në potencialet kimike.
Për fazën më të madhe: 
Për gazin në sipërfaqe: 
Në një gjendje ekuilibri  , d.m.th.
, d.m.th.

Në ekuacionin e Henrit konstanta nuk varet nga përqendrimi
Ekuacioni i Henrit është i vlefshëm në rajonin e presioneve dhe përqendrimeve të ulëta. Ndërsa përqendrimi rritet, 2 lloje devijimesh nga ligji i Henrit janë të mundshme:
1 – devijime pozitive, D zvogëlohet, A zvogëlohet
2 - devijime negative, D - rritet, A - rritet.
Lloji i devijimit përcaktohet nga mbizotërimi i një ose një lloji tjetër të ndërveprimit adsorbent-adsorbat.
Me ndërveprim të fortë ngjitës, koeficientët e aktivitetit rriten - një devijim pozitiv. Në rastin e ndërveprimeve kohezive, vërehen devijime negative.
Adsorbimi monomolekular.
Izotermia Langmuir.
Modelet më të thjeshta u morën në teorinë e Henrit. Langmuir propozoi një teori sipas së cilës adsorbimi konsiderohet si një reaksion kuazi-kimik. ku:
Sipërfaqja është energjikisht homogjene.
Adsorbimi është i lokalizuar, çdo qendër adsorbimi ndërvepron me një molekulë adsorbuese.
Molekulat e adsorbatit nuk ndërveprojnë me njëra-tjetrën.
Adsorbimi me një shtresë.

 - sipërfaqe,
- sipërfaqe,  - adsorbohen,
- adsorbohen,  - kompleksi i adsorbimit.
- kompleksi i adsorbimit.
 , pastaj përqendrimi i vendeve të adsorbimit:
, pastaj përqendrimi i vendeve të adsorbimit:  ,
, - duke kufizuar adsorbimin.
- duke kufizuar adsorbimin.
 , atëherë konstanta e reagimit është:
, atëherë konstanta e reagimit është: 

 - Ekuacioni Langmuir.
- Ekuacioni Langmuir.
Varësia e adsorbimit nga përqendrimi
1 )
)
 ,
,
2) zona me përqendrime të larta
 - duke kufizuar adsorbimin, formimi i një shtrese monomolekulare
- duke kufizuar adsorbimin, formimi i një shtrese monomolekulare
Për energjinë e Gibbs: .
g është faktori i entropisë.
Në rastin e izotermës së Henrit, energjia Gibbs karakterizon kalimin e adsorbatit nga gjendja standarde në masë në gjendjen standarde në sipërfaqe. Në rastin e izotermës Langmuir  karakterizon shkallën e afinitetit ndërmjet adsorbentit dhe adsorbatit.
karakterizon shkallën e afinitetit ndërmjet adsorbentit dhe adsorbatit.
 gjetur nga van't Hoff isobar.
gjetur nga van't Hoff isobar.

 , Pastaj
, Pastaj  , nga këtu
, nga këtu  .
.

 - shkalla e mbushjes së sipërfaqes.
- shkalla e mbushjes së sipërfaqes.




 - numri i vendeve të lira,
- numri i vendeve të lira,  - numri i vendeve të zëna.
- numri i vendeve të zëna.
 ,
,

ato. në rajonin e përqendrimeve të larta, numri i vendeve të lira është në përpjesëtim të zhdrejtë me sasinë e adsorbatit.
Adsorbimi i një përzierje gazesh në një sipërfaqe homogjene.
Në këtë rast, procesi i përthithjes konsiderohet si dy reaksione paralele.
 (1)
(1)

 (2)
(2)


Adsorbimi i një përzierje gazesh në një sipërfaqe jo uniforme.
Në rastin e një sipërfaqeje jo uniforme, nuk mund të kufizohet në mbushjet mesatare.
Si rezultat i konkurrencës, lokalizimi i adsorbateve të ndryshëm është i mundur në zona të llojeve të ndryshme.
Në këtë rast relacioni  .
.
 ,
,
 - presioni i avullit të ngopur të adsorbatit.
- presioni i avullit të ngopur të adsorbatit.
 ,
,
 - nxehtësia e përthithjes.
- nxehtësia e përthithjes.
"+" - varësia simbate, "-" - varësia antibate, "N" - pa korrelacion.
"+" - adsorbimi vazhdon sipas të njëjtit mekanizëm. Në zonat më të favorshme nga pikëpamja energjetike, thithet kryesisht gazi me afinitet të lartë për sipërfaqen.
“-” - adsorbimi ndodh përmes mekanizmave të ndryshëm dhe deri në një moment të caktuar kohor nuk ka konkurrencë për sipërfaqen.
Adsorbimi monomolekular realizohet kryesisht gjatë përthithjes fizike të gazeve në vlera të ulëta. fq, si dhe në ndërfaqen lëng/gaz.
Adsorbimi polimolekular.
Teoria BET(Brunauer, Emmett, Teller).
Në rastin kur formimi i një shtrese të vetme nuk mjafton për të kompensuar energjinë sipërfaqësore, adsorbimi është polimolekular dhe mund të konsiderohet si rezultat i kondensimit të detyruar nën veprimin e forcave sipërfaqësore.
Pikat kryesore:
Kur një molekulë adsorbate godet një vend të pushtuar, formohet një grup i shumëfishtë.
Ndërsa afrohemi fq te fq s zvogëlohet numri i vendeve të adsorbimit falas. Fillimisht shtohet dhe më pas zvogëlohet numri i vendeve që zënë teke, dyshe etj. në grupe.
Në fq =fq s adsorbimi kthehet në kondensim.
Nuk ka ndërveprime horizontale.
Për shtresën e parë plotësohet izotermia Langmuir.
Sipërfaqja konsiderohet si një grup vendesh adsorbimi. Vlen kushti i ekuilibrit dinamik: shpejtësia e kondensimit në vendet e lira është e barabartë me shpejtësinë e avullimit nga vendet e zëna.
a është koeficienti i kondensimit (fraksioni i molekulave të kondensuar në sipërfaqe);
 ,
,
Zm – numri maksimal i vendeve të lira.
 - frekuenca e dridhjeve atomike në drejtim pingul me sipërfaqen.
- frekuenca e dridhjeve atomike në drejtim pingul me sipërfaqen.
Për shtresën e parë, kushtet e ekuilibrit dinamik:



 , Pastaj
, Pastaj
 - Ekuacioni Langmuir.
- Ekuacioni Langmuir.
Për shtresën e dytë do të jetë e vërtetë: 
Për shtresën e i-të: 
Për thjeshtësi, supozohet se a dhe ν janë të njëjta për të gjitha shtresat përveç të parës. Për të gjitha shtresat, përveç të parës, nxehtësia e përthithjes është konstante. Për shtresën e fundit, nxehtësia e përthithjes është e barabartë me nxehtësinë e kondensimit. Si rezultat, u mor ekuacioni
 (*)
(*)
C- konstante,
Në rastin e teorisë BET, konstantja ME karakterizon energjinë Gibbs të adsorbimit të pastër. Ekuacioni përmban vetëm një konstante, dhe ky ekuacion është gjithashtu shumë i rëndësishëm për përcaktimin e sipërfaqes specifike të adsorbentit.
Meqenëse nxehtësia lirohet si rezultat i adsorbimit, sipërfaqet specifike përcaktohen në temperatura të ulëta.
 ????????????
????????????


E meta kryesore e teorisë– neglizhencë e ndërveprimeve horizontale në favor të atyre vertikale.
Ekuacioni qëndron në intervalin  nga 0.05 në 0.3.
nga 0.05 në 0.3.
Ku  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0.3 - ndërveprimi adsorbat - adsorbat është prekur.
> 0.3 - ndërveprimi adsorbat - adsorbat është prekur.
Kontabiliteti për ndërveprimet adsorbat-adsorbat.
Ndërveprimet ndodhin kur molekulat ose molekulat e degëzuara absorbohen në një sipërfaqe jopolare. Të aftë për të formuar bashkëpunëtorë. Në këtë rast, forma e izotermave të adsorbimit ndryshon.
A  adsorbenti nuk është polar.
adsorbenti nuk është polar.
Grafiku 1 korrespondon me ndërveprimet e dobëta adsorbat-adsorbat dhe ndërveprimet e forta adsorbat-adsorbues.
Grafiku 2 korrespondon me ndërveprimet e forta adsorbat-adsorbat dhe të fortë adsorbat-adsorbues.
Grafiku 3 korrespondon me ndërveprimin e fortë adsorbat-adsorbat dhe ndërveprimin e dobët adsorbat-adsorbues.
 ,
,

Në rastin e ndërveprimit midis molekulave të adsorbatit, është e nevojshme të merren parasysh ndryshimet në koeficientët e aktivitetit. Dhe ky ekuacion shkruhet si:
 - Ekuacioni Frunkin, Fowler, Guggenheim.
- Ekuacioni Frunkin, Fowler, Guggenheim.
k– konstante e tërheqjes.
Teoria potenciale e Polyany.
Kjo teori nuk nxjerr asnjë lloj izotermi adsorbimi, por bën të mundur llogaritjen e izotermave në një temperaturë të ndryshme.
Adsorbimi- ky është rezultat i tërheqjes së adsorbatit në sipërfaqen e adsorbentit për shkak të veprimit të potencialit të adsorbimit, i cili nuk varet nga prania e molekulave të tjera dhe varet nga distanca midis sipërfaqes dhe molekulës së adsorbatit.
 ,
,
 - potenciali i përthithjes.
- potenciali i përthithjes.
Meqenëse sipërfaqja është jo uniforme, distanca zëvendësohet nga vëllimi i përthithjes  .Vëllimi i përthithjesështë vëllimi i mbyllur midis sipërfaqes dhe pikës që i korrespondon një vlere të caktuar
.Vëllimi i përthithjesështë vëllimi i mbyllur midis sipërfaqes dhe pikës që i korrespondon një vlere të caktuar  .
.
Potenciali i adsorbimitështë puna e transferimit të 1 mol adsorbati jashtë një vëllimi të caktuar adsorbimi në një pikë të caktuar të vëllimit të adsorbimit (ose puna e transferimit të 1 mol avulli të ngopur të një adsorbati që është në ekuilibër me një adsorbat të lëngshëm në mungesë të një adsorbuesi në një fazë avulli në ekuilibër me adsorbentin).

Kurba karakteristike
 - potenciali i përthithjes,
- potenciali i përthithjes, 
Për një adsorbent të caktuar dhe adsorbate të ndryshme, sa vijon është e vërtetë: 
Për lloje të ndryshme adsorbatesh  ,
,
Ku  potencialet për izotermat e absorbimit në presione relative
potencialet për izotermat e absorbimit në presione relative  për adsorbatin 1 dhe për adsorbatin 2. Ky raport është një vlerë konstante.
për adsorbatin 1 dhe për adsorbatin 2. Ky raport është një vlerë konstante.
 - koeficienti i afinitetit
- koeficienti i afinitetit
Teoria e kondensimit kapilar.
Ecuria e procesit të adsorbimit varet kryesisht nga struktura e trupit poroz.
|
Mikroporoz | |
|
Poroze kalimtare | |
|
Makroporoz |
Në rastin e sorbenteve mikroporoze, fushat e forcave të adsorbimit mbivendosen. Në rastin e sorbentëve makroporozë, poret veprojnë si kanale transporti. Proceset e kondensimit janë më të rëndësishmet në trupat porozë në kalim. Kondensimi kapilar fillon në vlera të caktuara fq Dhe  , kur një pjesë e energjisë sipërfaqësore tashmë është kompensuar. Një kusht i domosdoshëm është që sipërfaqja të laget vetë. Procesi është përshkruar Ekuacioni Thompson–Kelvin.
, kur një pjesë e energjisë sipërfaqësore tashmë është kompensuar. Një kusht i domosdoshëm është që sipërfaqja të laget vetë. Procesi është përshkruar Ekuacioni Thompson–Kelvin.

 - për rastin e lagështimit, qendra e lakimit është në fazën e gazit.
- për rastin e lagështimit, qendra e lakimit është në fazën e gazit.
Në rastin e kondensimit kapilar, izotermia e adsorbimit ka një formë histerike. Dega e poshtme korrespondon me procesin e përthithjes, dhe dega e sipërme korrespondon me procesin e desorbimit.
Të gjitha llojet e poreve mund të reduktohen në tre lloje:
|
Konike |
Cilindrike me një fund të mbyllur |
Cilindrike me dy skaje të hapura |
|
Mbushja e procesit kryhet nga fundi i poreve. Izotermi i përthithjes dhe izotermi i desorbimit në këtë rast përkojnë, pasi procesi i përthithjes fillon nga një sferë dhe procesi i desorbimit gjithashtu fillon me zhdukjen e disa sferave.
↓ |
Nuk ka histerezë. Goditja e përparme dhe e kundërt përshkruhen nga ekuacioni:
|
Nuk ka fund askund, mbushja e poreve do të shkojë përgjatë mureve të cilindrit.
cilindër: Izotermia do të ketë një pamje histerike.
↓ |


 - sferë,
- sferë, ,
,



NË  Në kushte lagështie, kondensimi ndodh në presione më të ulëta, gjë që është energjikisht e favorshme. Nga dega e desorbimit fitohen kurbat e shpërndarjes së madhësisë së poreve.
Në kushte lagështie, kondensimi ndodh në presione më të ulëta, gjë që është energjikisht e favorshme. Nga dega e desorbimit fitohen kurbat e shpërndarjes së madhësisë së poreve.
Maksimumi i lakores diferenciale zhvendoset majtas në lidhje me pikën e lakimit të lakores integrale. Vëllimi i përgjithshëm i poreve të vogla është i vogël, por ka sipërfaqe të mëdha. Me rritjen e madhësisë së poreve, vëllimi i tyre rritet sa  , dhe zona eshte si
, dhe zona eshte si  , për shkak të kësaj, vërehet një zhvendosje në maksimumin e kurbës diferenciale.
, për shkak të kësaj, vërehet një zhvendosje në maksimumin e kurbës diferenciale.
Adsorbimi në ndërfaqen solid-lëng.
Në rastin e adsorbimit në ndërfaqen e gazit të ngurtë, ne neglizhuam një komponent. Në rastin e përthithjes në ndërfaqen solid-lëng, adsorbati zhvendos molekulat e tretësit nga sipërfaqja e adsorbentit.
 ,
,
Ekuacioni është i saktë:

 ,
,
N 1, N 2 – fraksionet mole të tretësit dhe përbërësit, N 1 + N 2 = 1, pastaj
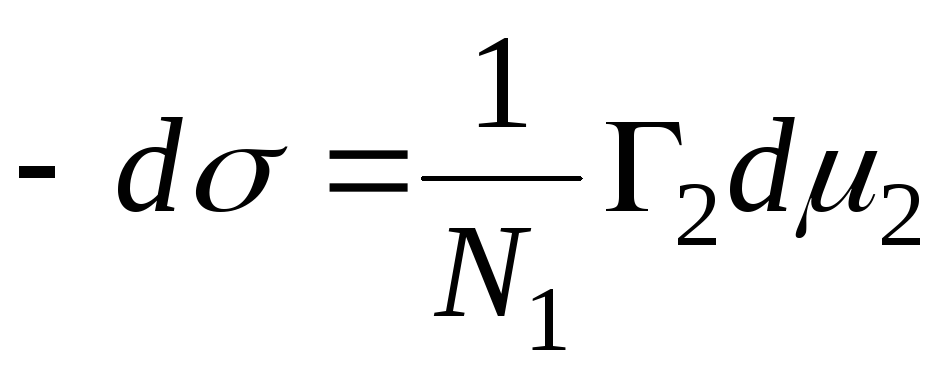 ,
=>
,
=>
 , atëherë është ekuacioni i adsorbimit për ndërfaqen solid-lëng.
, atëherë është ekuacioni i adsorbimit për ndërfaqen solid-lëng.
Adsorbimi (G) > 0 në  <
0
<
0
Nëse vlerat  sepse komponenti dhe tretësi janë shumë të ndryshme, në këtë rast varësia G nga N ka një ekstrem në vlerë N
~ 0,5.
sepse komponenti dhe tretësi janë shumë të ndryshme, në këtë rast varësia G nga N ka një ekstrem në vlerë N
~ 0,5.
E  nëse
nëse  kanë vlera të afërta, në këtë rast shenja e adsorbimit mund të ndryshojë. Varësia G nga N kalon boshtin x
kanë vlera të afërta, në këtë rast shenja e adsorbimit mund të ndryshojë. Varësia G nga N kalon boshtin x
Funksioni i pikës së kryqëzimit G(N) me boshtin x quhet azeotrope adsorbuese. Kjo do të thotë që të dy komponentët nuk mund të ndahen në një adsorbent të caktuar.
Ekuacioni i izotermës së përthithjes me konstante shkëmbimi.
Gjatë adsorbimit në ndërfaqen solid-lëng, ndodh një rishpërndarje konstante e përbërësve midis sipërfaqes së adsorbentit dhe vëllimit të tretësirës.
 - komponentët (- - referojuni sipërfaqes)
- komponentët (- - referojuni sipërfaqes)

 ,
,
 ,
, .
.
 ,
,


Adsorbimi në ndërfaqen lëng-gaz
R  Le të shqyrtojmë ndryshimin në profilin e përqendrimit kur kryqëzohet ndërfaqja lëng-gaz. Le të jetë komponenti 2 i paqëndrueshëm.
Le të shqyrtojmë ndryshimin në profilin e përqendrimit kur kryqëzohet ndërfaqja lëng-gaz. Le të jetë komponenti 2 i paqëndrueshëm.
Cs – përqendrimi në shtresën sipërfaqësore.
Bazuar në përkufizimin e adsorbimit të tepërt

Nëse komponenti nuk është i paqëndrueshëm, atëherë vlera e përthithjes do të shkruhet si më poshtë:
P  ri
ri 

Në barazimin.  natyra e një substance përshkruhet nga derivati i saj
natyra e një substance përshkruhet nga derivati i saj  .
.
Izotermi i tensionit sipërfaqësor mund të jetë i formës 1 ose 2:
1 – surfaktantë
2 – surfaktantë
Aktiviteti sipërfaqësor g është aftësia e substancave për të reduktuar tensionin sipërfaqësor në një sistem.

 - trashësia e shtresës sipërfaqësore
- trashësia e shtresës sipërfaqësore
C s– përqendrimi i përbërësit në shtresën sipërfaqësore
ME– përqendrimi i vëllimit
Për një seri homologe ekziston një rregull:
 - Rregulli i Traubo Duclos
- Rregulli i Traubo Duclos
Për një seri homologe, izotermi i adsorbimit duket kështu:
Në vend të A shkruajmë G, meqë adsorbimi është i tepruar në shtresën sipërfaqësore.
Izotermi i tensionit sipërfaqësor:

 - tensioni sipërfaqësor i një tretësi të pastër.
- tensioni sipërfaqësor i një tretësi të pastër.
 - ekuacioni themelor i adsorbimit;
- ekuacioni themelor i adsorbimit;
 - Ekuacioni Langmuir.
- Ekuacioni Langmuir.
Le t'i zgjidhim së bashku:

- ekuacioni Shishkovsky.
B– konstante për seritë homologe.
A- kur lëvizja nga një homolog në tjetrin rritet me 3-3,5 herë 
![]()
1 - zona me përqendrime të ulëta
![]()
2 – përqendrimi mesatar
3 – shtresa monomolekulare
Surfaktantët janë molekula difilike, d.m.th. përfshijnë një grup polar dhe një radikal hidrokarbur jopolar.
o është pjesa polare e molekulës.
| - pjesë jopolare e molekulës.
Në një tretës polar, molekulat e surfaktantit janë të orientuara në atë mënyrë që pjesa polare e molekulës përballet me tretësin, dhe pjesa jopolare të shtyhet në fazën e gazit.
Në ekuacionin e Shishkovskit  , është konstante për serinë homologjike.
, është konstante për serinë homologjike.
Efekti surfaktant fillon të shfaqet me n>5. Në përqendrime më të larta se përqendrimi i shtresës monomolekulare, micelizimi ndodh në tretësirat e surfaktantëve.
Micelle– quhet agregat i molekulave të surfaktantit amfifilë, radikalet hidrokarbure të të cilave formojnë një bërthamë dhe grupet polare kthehen në fazën ujore.
Masa micelale – masë micelale.
H  numri i molekulave - numri i grumbullimit.
numri i molekulave - numri i grumbullimit.
Micelat sferike
Në rastin e micelizimit vendoset ekuilibri në tretësirë
CMC – përqendrimi kritik i formimit të micelës.
Meqenëse ne e konsiderojmë micelën si një fazë të veçantë:
Për një seri homologjike ekziston një ekuacion empirik:
a– energjia e shpërbërjes së grupit funksional.
b – shtim i potencialit adsorbues, punë adsorbimi për njësi metilen.
– shtim i potencialit adsorbues, punë adsorbimi për njësi metilen.
Prania e një bërthame hidrokarbure në micele krijon mundësinë që komponimet që janë të patretshme në ujë të treten në tretësirat ujore të surfaktantëve; ky fenomen quhet tretje (ajo që tretet është tretësi, surfaktanti është tretësi).
Balta mund të jetë plotësisht jopolare, mund të përmbajë pjesë polare dhe jopolare dhe do të orientohet si një molekulë surfaktant.
Në çdo rast, gjatë tretjes ka një rritje të masës micellare dhe numrit të grumbullimit jo vetëm për shkak të përfshirjes së tretësirës, por edhe për shkak të rritjes së numrit të molekulave të surfaktantit të nevojshëm për të ruajtur një gjendje ekuilibri.
Tretësira është më efektive, sa më e ulët të jetë pesha molekulare e tretësirës.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
Efektiviteti i surfaktantëve varet nga vlera e CMC.
Presioni 2D i shtresës sipërfaqësore

→ -forcat e tensionit sipërfaqësor.
- presioni dydimensional.
Shtresa sipërfaqësore është një forcë e barabartë me diferencën në tensionin sipërfaqësor të një solucioni surfaktant dhe një tretës të pastër, të drejtuar drejt një sipërfaqeje të pastër.
Vendoset një ekuilibër midis tretësirës dhe shtresës sipërfaqësore



Në  ka një zonë ku
ka një zonë ku  varet në mënyrë lineare nga përqendrimi.
varet në mënyrë lineare nga përqendrimi.
G [mol/m2].
 -zona e zënë nga një mol i një lënde
-zona e zënë nga një mol i një lënde
Atëherë izotermi i presionit dydimensional do të ketë formën 
 - izotermi i presionit dydimensional.
- izotermi i presionit dydimensional.
Varësia  nga S M:
nga S M:

Në  - presioni dydimensional rritet ndjeshëm. Në
- presioni dydimensional rritet ndjeshëm. Në  dy-dimensionale është deformuar, duke shkaktuar rritje të papritur
dy-dimensionale është deformuar, duke shkaktuar rritje të papritur  .
.
Një film i kufizuar nga faza identike në të dy anët quhet i dyanshëm. Në filma të tillë vërehet lëvizje e vazhdueshme e pijeve amë.
Filmat me trashësi më të vogël se 5 nm quhen filma të zinj.

Shtresat e absorbimit duhet të kenë dy karakteristika: viskozitet dhe lëvizshmëri të lehtë, rrjedhshmëri dhe elasticitet.
Efekti Marangoni është vetë-shërues.
trekëndëshi i Gibbs,  - presioni i tepërt.
- presioni i tepërt.
Filmi është shtrirë dhe për faktin se një pjesë e lëngut është larguar, surfaktantët nxitojnë në hapësirën e lirë. Trekëndëshi i Gibbs.
Efekti i fuqisë së absorbimit të trupave.
Në sipërfaqen e filmit ka gjithmonë një shtresë adsorbimi, për të cilën më pas
Ekuacioni i Langmuir:


 në presion dydimensional
në presion dydimensional
 - një analog i ekuacionit Shishkovsky
- një analog i ekuacionit Shishkovsky
Dukuritë elektrokinetike. Shtresa elektrike e dyfishtë (EDL).
Modeli Gelemholtz. Teoria Gouy-Chapman.
1808 Fluturim

U – tub në formë, zhyt 2 elektroda në të. Ligji i enëve komunikuese shkelet dhe ndodh një ndryshim në nivelin e lëngut në tub - fenomene elektrokinetike.
Dukuritë kinetike:
Elektroforeza
Elektrozmoza
Potenciali i rrjedhës (rrjedhës).
Potenciali i sedimentimit
1 dhe 2 lindin kur aplikohet një diferencë potenciale; 3 dhe 4, grushtimi dhe sedimentimi i grimcave koloidale shkaktojnë shfaqjen e një ndryshimi potencial.
Elektrozmoza është lëvizja e një mjedisi dispersioni në lidhje me një fazë të disperzuar stacionare nën ndikimin e një rryme elektrike.
Elektroforeza - kjo është lëvizja e grimcave fazore të shpërndara në lidhje me një medium dispersioni të palëvizshëm nën ndikimin e një rryme elektrike.
P  Arsyeja e shfaqjes së dukurive elektrokinetike është ndarja hapësinore e ngarkesave dhe shfaqja e një shtrese elektrike të dyfishtë.
Arsyeja e shfaqjes së dukurive elektrokinetike është ndarja hapësinore e ngarkesave dhe shfaqja e një shtrese elektrike të dyfishtë.
Shtresa elektrike e dyfishtë është një kondensator i sheshtë, njëra pllakë formohet nga jonet që përcaktojnë potencialin, tjetra nga kundër-jonet. Jonet kontaminohen në të njëjtën mënyrë që ko-jonet përcaktuese të potencialit shtyhen në vëllimin e tretësirës. Distanca midis pllakave  . Potenciali bie në mënyrë lineare, diferenca potenciale
. Potenciali bie në mënyrë lineare, diferenca potenciale  .
.
Një ndryshim i jashtëm i potencialit shkakton shfaqjen e një moduli prerës  është një çift forcash për njësi sipërfaqe që veprojnë përgjatë sipërfaqes së një trupi të fortë.
është një çift forcash për njësi sipërfaqe që veprojnë përgjatë sipërfaqes së një trupi të fortë.

Në ekuilibër, moduli i prerjes është i barabartë me modulin e fërkimit viskoz (  ).
).

Në kushtet tona  ,
,

 - Ekuacioni Gelemholtz-Smalukowski
- Ekuacioni Gelemholtz-Smalukowski
 - shpejtësia lineare e zhvendosjes së fazës.
- shpejtësia lineare e zhvendosjes së fazës.
E– forca e fushës elektrike.
 - dallimi i mundshëm midis pllakave
- dallimi i mundshëm midis pllakave
 - lëvizshmëri elektroforetike [m 2 /(V*s)].
- lëvizshmëri elektroforetike [m 2 /(V*s)].
Modeli Helemholtz nuk merr parasysh lëvizjen termike të molekulave. Në realitet, shpërndarja e joneve në shtresën e dyfishtë është më komplekse.
Gui dhe Chapman identifikuan shkaqet e mëposhtme të DES:
Kalimi i një joni nga një fazë në tjetrën kur vendoset ekuilibri.
Jonizimi i lëndës së fazës së ngurtë.
Plotësimi i sipërfaqes me jone të pranishme në mjedisin e dispersionit.
Polarizimi nga një burim i jashtëm i rrymës.
Shtresa elektrike e dyfishtë ka një strukturë fuzzy ose difuze. Jonet priren të shpërndahen në mënyrë të barabartë në të gjithë shtresën difuze.
Shtresa difuze përbëhet nga kundërinone; gjatësia e shtresës përcaktohet nga energjia e tyre kinetike. Në temperaturat që i afrohen zeros absolute, kundër-jonet janë sa më afër që të jetë e mundur me jonet përcaktuese të potencialit.
Teoria e Danya bazohet në dy ekuacione:
ekuacioni Boltzmann

 - punojnë kundër forcave të bashkëveprimit elektrostatik.
- punojnë kundër forcave të bashkëveprimit elektrostatik.
 - dendësia e ngarkesës vëllimore.
- dendësia e ngarkesës vëllimore.

ekuacioni i Poisson-it 
Meqenëse trashësia e EDL është shumë më e vogël se madhësia e grimcave dhe për një EDL të sheshtë derivati në lidhje me koordinatat  Dhe
Dhe  është shfuqizuar.
është shfuqizuar. 
Për e y në y<<1 функцию можно разложить в ряд Маклорена:

Le të kufizohemi në dy terma të serisë, atëherë:


 - Trashësia DEL është distanca në të cilën potenciali DEL zvogëlohet e një herë.
- Trashësia DEL është distanca në të cilën potenciali DEL zvogëlohet e një herë.
Sa më e ulët të jetë temperatura, aq më pak  . Në T→0 – banesë DEL. Sa më i lartë të jetë përqendrimi, aq më shumë unë, aq më pak
. Në T→0 – banesë DEL. Sa më i lartë të jetë përqendrimi, aq më shumë unë, aq më pak  .
.





 “–” do të thotë që potenciali zvogëlohet me distancën. =>
“–” do të thotë që potenciali zvogëlohet me distancën. =>
=>

 ,
,
 - potenciali zvogëlohet në mënyrë eksponenciale.
- potenciali zvogëlohet në mënyrë eksponenciale.
Potenciali për densitetin e ngarkesës sipërfaqësore: 
Ngarkesa sipërfaqësore është një ngarkesë vëllimore me shenjën e kundërt, e integruar në distancë.




=>


Kur potenciali zvogëlohet me 2.7 herë - 
Kapaciteti me dy shtresa 
Disavantazhi i teorisë është se nuk merret parasysh prania e shtresës Helemholtz, d.m.th. nuk merr parasysh  , pra gabimet në përcaktimin e parametrave kryesorë. Gjithashtu nuk shpjegon ndikimin e joneve të natyrës së ndryshme në trashësinë e shtresës së dyfishtë elektrike.
, pra gabimet në përcaktimin e parametrave kryesorë. Gjithashtu nuk shpjegon ndikimin e joneve të natyrës së ndryshme në trashësinë e shtresës së dyfishtë elektrike.
teoria e Sternit. Struktura e micelës koloidale.
Shtresa elektrike e dyfishtë përbëhet nga dy pjesë: e dendur dhe difuze. Një shtresë e dendur formohet si rezultat i ndërveprimit të joneve të formimit të potencialit me ato të adsorbuara në mënyrë specifike. Këto jone, si rregull, janë pjesërisht ose plotësisht të dehidratuar dhe mund të kenë ngarkesë të njëjtë ose të kundërt me jonet që përcaktojnë potencialin. Varet nga raporti i energjisë së ndërveprimit elektrostatik  dhe potencialin specifik të adsorbimit
dhe potencialin specifik të adsorbimit  . Jonet e shtresës së dendur janë të fiksuara. Pjesa tjetër e joneve ndodhet në shtresën difuze; këto jone janë të lira dhe mund të lëvizin më thellë në tretësirë, d.m.th. nga një zonë me përqendrim më të lartë në një zonë me përqendrim më të ulët. Dendësia totale e ngarkesës përbëhet nga dy pjesë.
. Jonet e shtresës së dendur janë të fiksuara. Pjesa tjetër e joneve ndodhet në shtresën difuze; këto jone janë të lira dhe mund të lëvizin më thellë në tretësirë, d.m.th. nga një zonë me përqendrim më të lartë në një zonë me përqendrim më të ulët. Dendësia totale e ngarkesës përbëhet nga dy pjesë.

 -ngarkesa e shtreses Helmholtz
-ngarkesa e shtreses Helmholtz
 -Ngarkesa e shtresës difuze
-Ngarkesa e shtresës difuze
Sipërfaqja ka një numër të caktuar qendrash adsorbimi, secila prej të cilave ndërvepron me një kundërjon. Konstanta e një reaksioni të tillë pothuajse kimik është e barabartë me:

 , Ku
, Ku  - fraksioni mol i kundërjoneve në tretësirë
- fraksioni mol i kundërjoneve në tretësirë
Shpërndarja Helmholtz
Potenciali zvogëlohet në mënyrë lineare
Shpërndarja e potencialit gouy. Nuk ka shtresë të dendur, potenciali zvogëlohet në mënyrë eksponenciale nga vlera 
Shpërndarja e ashpër.
Fillimisht, ulja e potencialit është lineare dhe më pas eksponenciale.
Kur aplikohet një fushë elektrike në rastin e elektroforezës, nuk është grimca e fazës së ngurtë që lëviz drejtpërdrejt, por grimca e fazës së ngurtë me një shtresë jonesh që e rrethojnë atë. DES përsërit formën e grimcave të fazës së shpërndarë. Kur aplikohet një potencial, një pjesë e shtresës difuze shkëputet. Vija e thyerjes quhet kufiri rrëshqitës.
Potenciali që lind në kufirin e rrëshqitjes si rezultat i ndarjes së një pjese të shtresës difuze quhet potenciali elektrokinetik(Potenciali Zeta  ).
).
Një grimcë fazore e shpërndarë me një shtresë rrethuese kundërjonesh dhe një shtresë elektrike të dyfishtë quhet micelë.
Rregullat për shkrimin e micelave koloidale:


1-1 elektrolit karikues
T - grimca fazore e shpërndarë.
AA është kufiri midis pjesëve të dendura dhe difuze.
BB - kufiri rrëshqitës.
Kufiri rrëshqitës mund ose nuk mund të përkojë me linjën AA.
Vlera e pH në të cilën potenciali zeta është zero quhet pika izoelektrike.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2 NaCl
1. CaCl i tepërt 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - shënimi i micelës.
CaSO 4 m – agregat.
CaSO 4 m∙nCa 2+ – bërthama.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - grimcë.
2. Teprica e Na 2 SO 4
Na 2 SO 4 ↔2 Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelë
CaSO 4 m – agregat.
CaSO 4 m∙nSO 4 2 + – bërthama.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - grimca
Ekuacioni Gelemholtz-Smoluchowski

 - shpejtësia lineare e zhvendosjes së kufirit (në elektroosmozë).
- shpejtësia lineare e zhvendosjes së kufirit (në elektroosmozë).
 - dallimi i potencialit nëpër pllakat e kondensatorit (në elektroosmozë).
- dallimi i potencialit nëpër pllakat e kondensatorit (në elektroosmozë).


 - shpejtësia vëllimore e rrjedhës së tretësirës, S- zona e prerjes tërthore të qelizës.
- shpejtësia vëllimore e rrjedhës së tretësirës, S- zona e prerjes tërthore të qelizës.

E– forca e fushës elektrike.


 (për elektroosmozë).
(për elektroosmozë).
Për potencialin e rrjedhës:

 - potencial
- potencial
 - presioni në membranë
- presioni në membranë
Si rregull, vlerat e lëvizshmërisë elektroforetike dhe lëvizshmërisë elektroosmotike janë më të vogla se ato të llogaritura. Kjo ndodh për shkak të:
Efekti i relaksimit (kur një grimcë fazore e shpërndarë lëviz, simetria e atmosferës jonike prishet).
Frenimi elektroforetik (shfaqja e fërkimit shtesë si rezultat i lëvizjes së kundërjoneve).
Shtrembërimi i linjave të rrymës në rastin e grimcave përçuese elektrike.
Marrëdhënia midis tensionit sipërfaqësor dhe potencialit. ekuacioni Lippmann.
Formimi i EDL ndodh në mënyrë spontane për shkak të dëshirës së sistemit për të zvogëluar energjinë e tij sipërfaqësore. Në kushte konstante T Dhe fq ekuacioni i përgjithësuar i ligjit të parë dhe të dytë të termodinamikës duket si:
 (2)
(2)
(3), (1)=(3) =>
=>

 - Ekuacioni i parë i Lippmann-it.
- Ekuacioni i parë i Lippmann-it.
 - dendësia e ngarkesës sipërfaqësore.
- dendësia e ngarkesës sipërfaqësore.
 - kapaciteti diferencial.
- kapaciteti diferencial.
 - Ekuacioni i 2-të i Lippmann-it.
- Ekuacioni i 2-të i Lippmann-it.
ME– kapaciteti.
Le të zgjidhim ekuacionin e parë të Lippmann dhe ekuacionin themelor të adsorbimit:
 ,
,


 , Pastaj
, Pastaj
 - Ekuacioni Nernst
- Ekuacioni Nernst
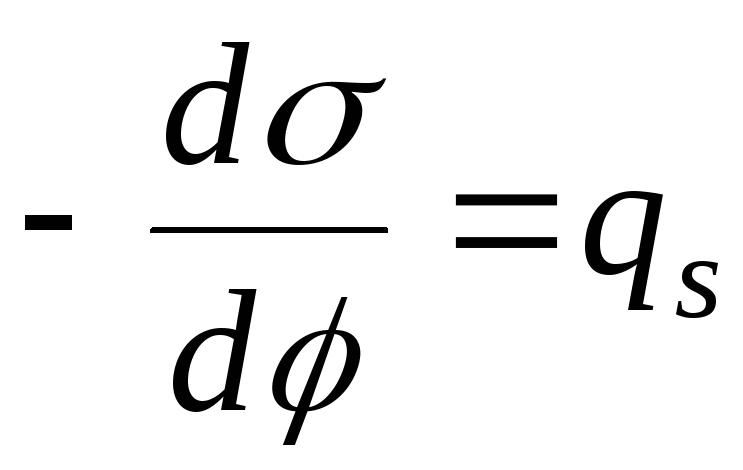 ,
,
 ,
,




 - ekuacioni i lakores elektrokapilare (ECC).
- ekuacioni i lakores elektrokapilare (ECC).
NË  :
: , Por
, Por 
Surfaktantët kationikë (CPAS) reduktojnë degën katodike të EKC.
Surfaktantët anionikë (APS) reduktojnë degën anodike të EKC.
Surfaktantët jooninik (NSAS) reduktojnë pjesën e mesme të ECC.
Stabiliteti i sistemeve të shpërndara. Presion shpërbërës.
Sistemet e shpërndara mund të ndahen:
Sistemet që janë termodinamikisht të paqëndrueshëm mund të jenë kinetikisht të qëndrueshëm për shkak të kalimit në një gjendje metastabile.
Ekzistojnë dy lloje të stabilitetit:
Stabiliteti i sedimentimit (në raport me gravitetin).
Stabiliteti agregativ. (në lidhje me ngjitjen)
Koagulimiështë një proces i ngjitjes së grimcave, që çon në humbjen e stabilitetit të grumbullimit. Koagulimi mund të shkaktohet nga ndryshimet e temperaturës, pH, trazimi dhe ultratingulli.
Koagulimi dallohet:
E kthyeshme.
E pakthyeshme.
Koagulimi ndodh me futjen e elektroliteve.
Rregullat e koagulimit:

Film- kjo është pjesa e sistemit që ndodhet midis dy sipërfaqeve ndërfaqe.
Presion shpërbërës ndodh kur trashësia e filmit zvogëlohet ndjeshëm si rezultat i ndërveprimit të shtresave sipërfaqësore që afrohen.

"-" - ndërsa trashësia e filmit zvogëlohet, presioni i shkëputjes rritet.
P 0 është presioni në fazën pjesa më e madhe, e cila është një vazhdimësi e ndërshtresës.
P 1 - presioni në film.
Teoria e stabilitetit. DLFO (Deryagin, Landau, Fairway, Overbeck).
Sipas teorisë DLFO, presioni i shkëputur ka dy komponentë:
Elektrostatike P E (pozitive, është për shkak të forcave të sprapsjes elektrostatike). Korrespondon me një ulje të energjisë Gibbs me rritjen e trashësisë së filmit.
molekulare P M (negativ, për shkak të veprimit të forcave tërheqëse). Shkaktohet nga ngjeshja e filmit për shkak të forcave kimike të sipërfaqes, rrezja e veprimit të forcave është të dhjetat e nm me një energji prej rreth 400 kJ/mol.
Energjia totale e ndërveprimit:

 - sistemi është totalisht i qëndrueshëm
- sistemi është totalisht i qëndrueshëm
 - sistem i paqëndrueshëm
- sistem i paqëndrueshëm
P  komponent pozitiv.
komponent pozitiv.
Rritja është për shkak të rritjes së energjisë potenciale kur ngjeshen filmat e hollë. Për filmat me trashësi të madhe, energjia e tepërt e joneve kompensohet dhe është e barabartë me ndërveprimin e energjisë në vëllimin e mediumit të shpërndarjes.
Nëse  (
( - trashësia e filmit,
- trashësia e filmit,  - rreze jonike) hollimi i filmit çon në zhdukjen dhe reduktimin e molekulave dhe joneve me energji minimale sipërfaqësore në të. Numri i grimcave fqinje zvogëlohet, si rezultat i së cilës rritet energjia potenciale e grimcave që mbeten në film.
- rreze jonike) hollimi i filmit çon në zhdukjen dhe reduktimin e molekulave dhe joneve me energji minimale sipërfaqësore në të. Numri i grimcave fqinje zvogëlohet, si rezultat i së cilës rritet energjia potenciale e grimcave që mbeten në film.


Teoria DLVO e konsideron bashkëveprimin e grimcave si bashkëveprim të pllakave.

Grimcat nuk ndërveprojnë



 - Ekuacioni i Laplasit,
- Ekuacioni i Laplasit,  ,
,


Për sipërfaqet me ngarkesë të dobët
Për sipërfaqe shumë të ngarkuara:

Komponenti molekular është bashkëveprimi i dy atomeve:
 ~
~
Ndërveprimi i një atomi me një sipërfaqe:

Le të marrim dy regjistrime:
D  Për të marrë përbërësin molekular, është e nevojshme të përmblidhen të gjitha energjitë e ndërveprimit të atomeve të pllakave të djathta dhe të majta.
Për të marrë përbërësin molekular, është e nevojshme të përmblidhen të gjitha energjitë e ndërveprimit të atomeve të pllakave të djathta dhe të majta.
Ku  - Konstanta Hamaker (merr parasysh natyrën e trupave ndërveprues).
- Konstanta Hamaker (merr parasysh natyrën e trupave ndërveprues).
Se. energjia e ndërveprimit të grimcave në një sistem mund të shprehet duke përdorur kthesat e mundshme.
I – minimumi i mundshëm primar. Kjo është një zonë e koagulimit të pakthyeshëm, forcat e tërheqjes mbizotërojnë.
II - zona e stabilitetit agregativ, mbizotërojnë forcat repulsive.
III – minimumi i potencialit dytësor (ose zona e flokulimit). Ekziston një shtresë elektrolitike midis grimcave të fazës së shpërndarë, dhe grimcat mund të ndahen dhe transferohen në zonën e qëndrueshmërisë së grumbullimit.

Kurba 1 - sistemi është totalisht i qëndrueshëm.
Kurba 2 – e qëndrueshme në zonën I, e paqëndrueshme në zonën II.
Kurba 3 - ka ndodhur koagulimi në sistem.
Lakorja 4 – në pikën 4 energjia totale e bashkëveprimit U=0,  , kjo pikë ekstreme korrespondon me fillimin e koagulimit të shpejtë.
, kjo pikë ekstreme korrespondon me fillimin e koagulimit të shpejtë.
Janë dy raste:
1. Sipërfaqe pak të ngarkuara:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - kjo është trashësia e shtresës që korrespondon me fillimin e procesit të koagulimit.
- kjo është trashësia e shtresës që korrespondon me fillimin e procesit të koagulimit.






 - për sipërfaqe me ngarkesë të dobët
- për sipërfaqe me ngarkesë të dobët
 Pastaj
Pastaj 
2. Për sipërfaqe shumë të ngarkuara:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Le të vendosim në katror (3)

Koagulimi:
Në adsorbimin specifik, jonet mund të absorbohen në sasi super-ekuivalente në mënyrë që sipërfaqja të mund të ndryshojë ngarkesën e saj. Sipërfaqja është e rimbushur.
Në rastin e adsorbimit specifik, mund të absorbohen jo vetëm të shenjave të kundërta, por edhe të së njëjtës shenjë.
Nëse jonet e së njëjtës shenjë si sipërfaqja absorbohen, atëherë në shtresën sipërfaqësore nuk do të ketë një rënie të potencialit, por një rritje në të.

Koagulimi i neutralizimit (ndodh me pjesëmarrjen e grimcave të ngarkuara dobët dhe varet jo vetëm nga ngarkesa e elektrolitit-koagulator, por edhe nga potenciali në kufirin e shtresave të dendura dhe difuze).
Teoria e koagulimit të shpejtë të Smoluchowski.

Varësia e shkallës së koagulimit nga përqendrimi i elektrolitit.
I - shkalla e koagulimit është e ulët,
II - shkalla e koagulimit është pothuajse proporcionale me përqendrimin e elektrolitit.
III - rajoni i koagulimit të shpejtë, shpejtësia është praktikisht e pavarur nga përqendrimi.
Dispozitat themelore:
Sol fillestar është monodisperse, grimca të ngjashme kanë një formë sferike.
Të gjitha përplasjet e grimcave janë efektive.
Kur dy grimca primare përplasen, formohet një grimcë dytësore. Sekondar + parësor = terciar. primare, dytësore, terciare - shumësi.
Për sa i përket kinetikës kimike, procesi i koagulimit mund të përshkruhet nga ekuacioni:
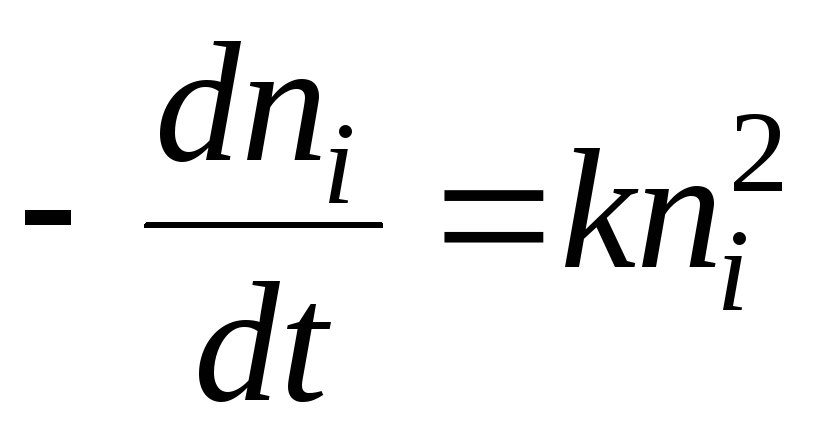
Zgjidhja do të jetë ekuacioni: 
 - gjysma e kohës së koagulimit. Kjo është koha gjatë së cilës numri i grimcave të sollit zvogëlohet me 2 herë.
- gjysma e kohës së koagulimit. Kjo është koha gjatë së cilës numri i grimcave të sollit zvogëlohet me 2 herë.

 ,
,
 ,
,
 ,
,


Me rritjen e shumëfishimit, maksimumi i kurbave të koagulimit zhvendoset drejt vlerave më të mëdha  .
.
Të metat:
Supozimi i monodispersitetit.
Supozimi për efektivitetin e të gjitha përplasjeve.


