In the case of interaction between two atoms:
U – interaction energy;
U = U PRIOR. + U RETURN
 - Lennard-Jones equation
, c, b, m = const
- Lennard-Jones equation
, c, b, m = const
In cases of interaction of atoms with a solid surface, it is necessary to sum up all interactions.
x – distance to surface
r – radius of action of attractive forces
dV – volume
n – number of surface molecules
U ADS. – energy of adsorption interaction



In the case of adsorption, the attraction increases. And in the case of nonpolar-nonpolar interaction, adsorption is predominantly localized in recesses.
Electrostatic interaction.
Polar adsorbent – non-polar adsorbate
Non-polar adsorbent – polar adsorbate
Polar adsorbent – polar adsorbate.
M  The adsorbate molecule is represented as a dipole, and the adsorbent is represented as a conductor in which the adsorbate molecule induces a dipole mirror symmetrically with respect to the given one.
The adsorbate molecule is represented as a dipole, and the adsorbent is represented as a conductor in which the adsorbate molecule induces a dipole mirror symmetrically with respect to the given one.
X – distance to the middle
When interacting, potential arises:
 ,
,
 - dipole moment.
- dipole moment.
The potential tends to take on the maximum value, i.e. dipoles tend to orient themselves perpendicular to the surface.
Since an increase in temperature promotes the growth of Brownian motion, it leads to inhibition of the adsorption process.
In the case of electrostatic interaction, the adsorbate is predominantly localized on the protrusions.
Fundamental adsorption equation.
In the case of adsorption, a redistribution of the component occurs, which means the chemical potential changes. The adsorption process can be considered as the transition of surface energy into chemical energy.
Layer volume = 0, then the generalized equation of the I and II laws of thermodynamics:
T = const; (1) = (2) => 

For a two-component system:
,
 ,
,
 =>
=>
=>
 - Gibbs adsorption equation
.
- Gibbs adsorption equation
.
For the case of TV adsorption. body - gas: , 
 ,
,

 - isotherm
- isotherm
 - isobar
- isobar
 - isopycnal
- isopycnal
 - isostere
- isostere
Isotherm, isopycne, isostere are related to each other.
Because adsorption function 



Henry isotherm Langmuir isotherm



Thermodynamics. Adsorption.
For condensed matter:
 ,
,
 ,
,
 - integral change in Gibbs energy
.
- integral change in Gibbs energy
.

P – pressure over a curved surface, Р S – pressure over a flat surface
 - adsorption potential
- adsorption potential
Differential change in entrapy 
 , Г = const
, Г = const
- differential entropy change
- differential enthalpy of adsorption
 - isosteric heat of adsorption
- isosteric heat of adsorption
 - heat of condensation
- heat of condensation
 - net heat of adsorption
- net heat of adsorption

 ,
,




Qa – integral heat of adsorption, 
Qra – integral net heat of adsorption,

Henry's equation
The study of adsorption is complicated by the heterogeneity of the surface, so the simplest laws are obtained for homogeneous surfaces.
Let us consider the interaction of gases with a solid surface, when a gas transitions from an equilibrium state in the volume to an equilibrium state on the surface. This case is analogous to the equilibrium of gases in a gravity field.
 ,
,
 ,
=>
,
=> -Henry's equation
-Henry's equation
 - distribution coefficient
- distribution coefficient
During the adsorption process, a change in chemical potentials occurs.
For the bulk phase: 
For gas on the surface: 
In a state of balance  , i.e.
, i.e.

In Henry's equation the constant does not depend on concentration
Henry's equation is valid in the region of low pressures and concentrations. As concentration increases, 2 types of deviations from Henry’s law are possible:
1 – positive deviations, D decreases, A decreases
2 – negative deviations, D – increases, A – increases.
The type of deviation is determined by the predominance of one or another type of adsorbent-adsorbate interaction.
With strong adhesive interaction, the activity coefficients increase - a positive deviation. In the case of cohesive interactions, negative deviations are observed.
Monomolecular adsorption.
Langmuir isotherm.
The simplest patterns were obtained in Henry's theory. Langmuir proposed a theory according to which adsorption is considered as a quasi-chemical reaction. Wherein:
The surface is energetically homogeneous.
Adsorption is localized, each adsorption center interacts with one adsorbate molecule.
Adsorbate molecules do not interact with each other.
Monolayer adsorption.

 - surface,
- surface,  - adsorbate,
- adsorbate,  - adsorption complex.
- adsorption complex.
 , then the concentration of adsorption sites:
, then the concentration of adsorption sites:  ,
, - limiting adsorption.
- limiting adsorption.
 , then the reaction constant is:
, then the reaction constant is: 

 - Langmuir equation.
- Langmuir equation.
Dependence of adsorption on concentration
1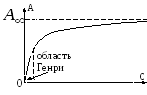 )
)
 ,
,

2) area of high concentrations
 - limiting adsorption, formation of a monomolecular layer
- limiting adsorption, formation of a monomolecular layer
For Gibbs energy: .
g is the entropy factor.
In the case of the Henry isotherm, the Gibbs energy characterizes the transition of the adsorbate from the standard state in the bulk to the standard state on the surface. In the case of the Langmuir isotherm  characterizes the degree of affinity between the adsorbent and the adsorbate.
characterizes the degree of affinity between the adsorbent and the adsorbate.
 found from the van't Hoff isobar.
found from the van't Hoff isobar.

 , Then
, Then  , from here
, from here  .
.

 - degree of surface filling.
- degree of surface filling.




 - number of free seats,
- number of free seats,  - number of occupied places.
- number of occupied places.
 ,
,

Those. in the region of high concentrations, the number of free sites is inversely proportional to the amount of adsorbate.
Adsorption of a mixture of gases on a homogeneous surface.
In this case, the adsorption process is considered as two parallel reactions.
 (1)
(1)

 (2)
(2)


Adsorption of a mixture of gases on a non-uniform surface.
In the case of a non-uniform surface, one cannot limit oneself to average fillings.
As a result of competition, localization of different adsorbates is possible in areas of different types.
In this case the relation  .
.
 ,
,
 - saturated vapor pressure of the adsorbate.
- saturated vapor pressure of the adsorbate.
 ,
,
 - heat of adsorption.
- heat of adsorption.
“+” - symbate dependence, “-” - antibate dependence, “N” - no correlation.
“+” - adsorption proceeds according to the same mechanism. In the most energetically favorable areas, gas with a high affinity for the surface is predominantly adsorbed.
“-” - adsorption occurs through various mechanisms and until a certain point in time there is no competition for the surface.
Monomolecular adsorption is predominantly realized during the physical adsorption of gases at low values p, as well as at the liquid/gas interface.
Polymolecular adsorption.
BET theory(Brunauer, Emmett, Teller).
In the case where the formation of a monolayer is not enough to compensate for the surface energy, adsorption is polymolecular and can be considered as a result of forced condensation under the action of surface forces.
Key points:
When an adsorbate molecule hits an occupied site, a multiple set is formed.
As we get closer p To p s the number of free adsorption sites decreases. Initially, the number of places occupied by singles, doubles, etc. increases and then decreases. in sets.
At p =p s adsorption turns into condensation.
There are no horizontal interactions.
For the first layer, the Langmuir isotherm is fulfilled.
The surface is considered as a set of adsorption sites. The condition of dynamic equilibrium is valid: the rate of condensation in free places is equal to the rate of evaporation from occupied places.
a is the condensation coefficient (the fraction of molecules condensed on the surface);
 ,
,
Zm – maximum number of free seats.
 - frequency of atomic vibrations in the direction perpendicular to the surface.
- frequency of atomic vibrations in the direction perpendicular to the surface.
For the first layer, dynamic equilibrium conditions:



 , Then
, Then
 - Langmuir equation.
- Langmuir equation.
For the second layer it will be true: 
For the i-th layer: 
For simplicity, it is assumed that a and ν are the same for all layers except the first. For all layers except the first, the heat of adsorption is constant. For the last layer, the heat of adsorption is equal to the heat of condensation. As a result, the equation was obtained
 (*)
(*)
C– constant,
In the case of BET theory, the constant WITH characterizes the Gibbs energy of pure adsorption. The equation contains only one constant, and this equation is also very important for determining the specific surface area of the adsorbent.
Since heat is released as a result of adsorption, specific surface areas are determined at low temperatures.
 ????????????
????????????


The main drawback of the theory– neglect of horizontal interactions in favor of vertical ones.
The equation holds in the range  from 0.05 to 0.3.
from 0.05 to 0.3.
Where  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0.3 – the adsorbate – adsorbate interaction is affected.
> 0.3 – the adsorbate – adsorbate interaction is affected.
Accounting for adsorbate-adsorbate interactions.
Interactions occur when branched molecules or molecules are adsorbed on a nonpolar surface. Capable of forming associates. In this case, the shape of the adsorption isotherms changes.
A  the adsorbent is not polar.
the adsorbent is not polar.
Graph 1 corresponds to weak adsorbate-adsorbate interactions and strong adsorbate-adsorbent interactions.
Graph 2 corresponds to strong adsorbate-adsorbate and strong adsorbate-adsorbent interactions.
Graph 3 corresponds to strong adsorbate-adsorbate interaction and weak adsorbate-adsorbent interaction.
 ,
,

In the case of interaction between adsorbate molecules, it is necessary to take into account changes in activity coefficients. And this equation is written as:
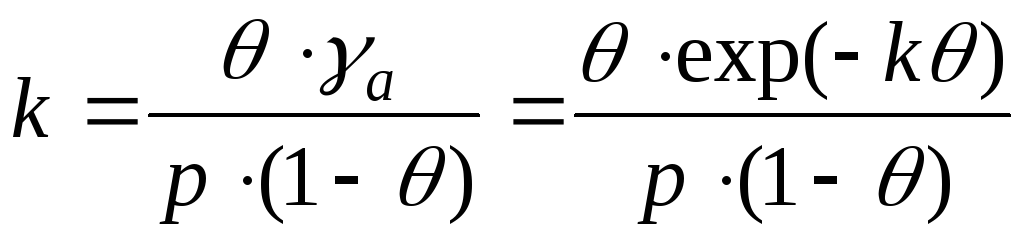 - Frunkin, Fowler, Guggenheim equation.
- Frunkin, Fowler, Guggenheim equation.
k– attraction constant.
Polyany's potential theory.
This theory does not derive any type of adsorption isotherm, but makes it possible to calculate isotherms at a different temperature.
Adsorption- this is the result of the attraction of the adsorbate to the surface of the adsorbent due to the action of the adsorption potential, which does not depend on the presence of other molecules and depends on the distance between the surface and the adsorbate molecule.
 ,
,
 - adsorption potential.
- adsorption potential.
Since the surface is non-uniform, the distance is replaced by the adsorption volume  .Adsorption volume is the volume enclosed between the surface and the point corresponding to a given value
.Adsorption volume is the volume enclosed between the surface and the point corresponding to a given value  .
.
Adsorption potential is the work of transferring 1 mole of adsorbate outside a given adsorption volume to a given point of the adsorption volume (or the work of transferring 1 mole of saturated vapor of an adsorbate that is in equilibrium with a liquid adsorbate in the absence of an adsorbent into a vapor phase in equilibrium with the adsorbent).

Characteristic curve
 - adsorption potential,
- adsorption potential, 
For a given adsorbent and various adsorbates, the following is true: 
For different types of adsorbates  ,
,
Where  potentials for adsorption isotherms at relative pressures
potentials for adsorption isotherms at relative pressures  for adsorbate 1 and for adsorbate 2. This ratio is a constant value.
for adsorbate 1 and for adsorbate 2. This ratio is a constant value.
 - affinity coefficient
- affinity coefficient
Theory of capillary condensation.
The course of the adsorption process largely depends on the structure of the porous body.
|
Microporous | |
|
Transitional porous | |
|
Macroporous |
In the case of microporous sorbents, the fields of adsorption forces overlap. In the case of macroporous sorbents, pores act as transport channels. Condensation processes are most significant in transitionally porous bodies. Capillary condensation begins at certain values p And  , when part of the surface energy has already been compensated. A necessary condition is that the surface must be self-wetting. The process is described Thompson–Kelvin equation.
, when part of the surface energy has already been compensated. A necessary condition is that the surface must be self-wetting. The process is described Thompson–Kelvin equation.

 - for the case of wetting, the center of curvature is in the gas phase.
- for the case of wetting, the center of curvature is in the gas phase.
In the case of capillary condensation, the adsorption isotherm has a hysteretic form. The lower branch corresponds to the adsorption process, and the upper branch corresponds to the desorption process.
All types of pores can be reduced to three types:
|
Conical |
Cylindrical with one closed end |
Cylindrical with two open ends |
|
Process filling is carried out from the bottom of the pore. The adsorption isotherm and the desorption isotherm in this case coincide, since the adsorption process begins from a sphere and the desorption process also begins with the disappearance of some spheres.
↓ |
There is no hysteresis. Forward and reverse stroke are described by the equation:
|
There is no bottom anywhere, the filling of the pore will go along the walls of the cylinder.
cylinder: The isotherm will have a hysteretic appearance.
↓ |


 - sphere,
- sphere, ,
,



IN  Under wetting conditions, condensation occurs at lower pressures, which is energetically favorable. From the desorption branch, pore size distribution curves are obtained.
Under wetting conditions, condensation occurs at lower pressures, which is energetically favorable. From the desorption branch, pore size distribution curves are obtained.
The maximum of the differential curve is shifted to the left relative to the inflection point of the integral curve. The total volume of small pores is small, but has large surface areas. With increasing pore size, their volume increases as  , and the area is like
, and the area is like  , due to this, a shift in the maximum of the differential curve is observed.
, due to this, a shift in the maximum of the differential curve is observed.
Adsorption at the solid-liquid interface.
In the case of adsorption at the solid-gas interface, we neglected one component. In the case of adsorption at the solid-liquid interface, the adsorbate displaces solvent molecules from the surface of the adsorbent.
 ,
,
The equation is correct:

 ,
,
N 1, N 2 – mole fractions of solvent and component, N 1 + N 2 = 1, then
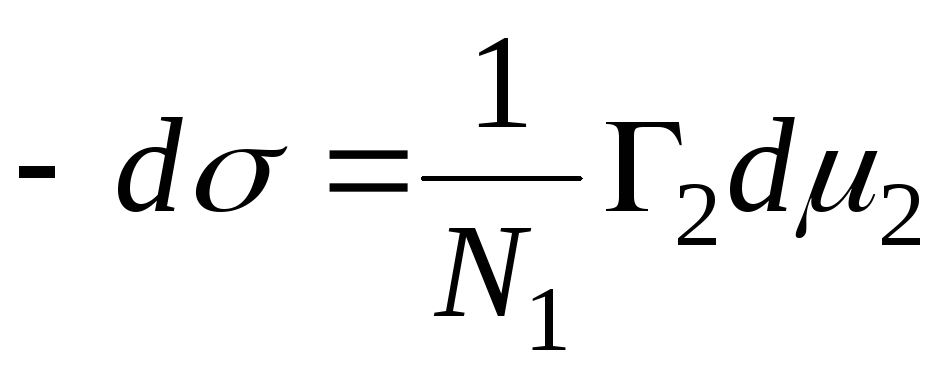 ,
=>
,
=>
 , then is the adsorption equation for the solid-liquid interface.
, then is the adsorption equation for the solid-liquid interface.
Adsorption (G) > 0 at  <
0
<
0
If the values  for the component and the solvent are very different, in this case the dependence G from N has an extremum at the value N
~ 0,5.
for the component and the solvent are very different, in this case the dependence G from N has an extremum at the value N
~ 0,5.
E  if
if  have close values, in this case the sign of adsorption may change. Addiction G from N crosses the x-axis
have close values, in this case the sign of adsorption may change. Addiction G from N crosses the x-axis
Function intersection point G(N) with the x-axis is called adsorption azeotrope. This means that the two components cannot be separated on a given adsorbent.
Equation of adsorption isotherm with exchange constant.
During adsorption at the solid-liquid interface, a constant redistribution of components occurs between the surface of the adsorbent and the volume of the solution.
 - components (- - refer to the surface)
- components (- - refer to the surface)

 ,
,
 ,
, .
.
 ,
,


Adsorption at the liquid-gas interface
R  Let us consider the change in the concentration profile as the liquid-gas interface is crossed. Let component 2 be volatile.
Let us consider the change in the concentration profile as the liquid-gas interface is crossed. Let component 2 be volatile.
Cs – concentration in the surface layer.
Based on the definition of excess adsorption

If the component is not volatile, then the adsorption value will be written as follows:
P  ri
ri 

In Eq.  the nature of a substance is described by its derivative
the nature of a substance is described by its derivative  .
.
The surface tension isotherm can be of the form 1 or 2:
1 – surfactants
2 – surfactants
Surface activity g is the ability of substances to reduce surface tension in a system.

 - thickness of the surface layer
- thickness of the surface layer
C s– concentration of the component in the surface layer
WITH– volume concentration
For a homologous series there is a rule:
 - Traubo Duclos rule
- Traubo Duclos rule
For a homologous series, the adsorption isotherm looks like this:
Instead of A we write G, since adsorption is excessive in the surface layer.
Surface tension isotherm:

 - surface tension of a pure solvent.
- surface tension of a pure solvent.
 - fundamental adsorption equation;
- fundamental adsorption equation;
 - Langmuir equation.
- Langmuir equation.
Let's solve them together:

- Shishkovsky equation.
B– constant for the homologous series.
A- when moving from one homolog to another increases by 3-3.5 times 
![]()
1 – area of low concentrations
![]()
2 – average concentration
3 – monomolecular layer
Surfactants are diphilic molecules, i.e. include a polar group and a non-polar hydrocarbon radical.
o is the polar part of the molecule.
| - non-polar part of the molecule.
In a polar solvent, surfactant molecules are oriented in such a way that the polar part of the molecule faces the solvent, and the non-polar part is pushed into the gas phase.
In Shishkovsky's equation  , it is constant for the homological series.
, it is constant for the homological series.
The surfactant effect begins to appear with n>5. At concentrations higher than the concentration of the monomolecular layer, micellization occurs in surfactant solutions.
Micelle– is called an aggregate of amphiphilic surfactant molecules, the hydrocarbon radicals of which form a core, and the polar groups are turned into the aqueous phase.
Micelle mass – micellal mass.
H  number of molecules – aggregation number.
number of molecules – aggregation number.
Spherical micelles
In the case of micellization, equilibrium is established in the solution
CMC – critical concentration of micelle formation.
Since we consider the micelle to be a separate phase:
For a homological series there is an empirical equation:
a– energy of dissolution of the functional group.
b – increment of adsorption potential, adsorption work per methylene unit.
– increment of adsorption potential, adsorption work per methylene unit.
The presence of a hydrocarbon core in micelles creates the opportunity for compounds that are insoluble in water to be dissolved in aqueous solutions of surfactants; this phenomenon is called solubilization (what dissolves is the solubilisate, the surfactant is the solubilizer).
The mud may be completely non-polar, may contain both polar and non-polar parts and will be oriented like a surfactant molecule.
In any case, during solubilization there is an increase in micellar mass and aggregation number not only due to the inclusion of solubilizate, but also due to an increase in the number of surfactant molecules necessary to maintain an equilibrium state.
Solubilization is more effective, the lower the molecular weight of the solubilizate.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
The effectiveness of surfactants depends on the CMC value.
2D Surface Layer Pressure

→ -surface tension forces.
- two-dimensional pressure.
The surface layer is a force equal to the difference in surface tension of a surfactant solution and a pure solvent, directed towards a clean surface.
An equilibrium is established between the solution and the surface layer



At  there is an area where
there is an area where  depends linearly on concentration.
depends linearly on concentration.
G [mol/m2].
 -area occupied by one mole of a substance
-area occupied by one mole of a substance
Then the two-dimensional pressure isotherm will have the form 
 - two-dimensional pressure isotherm.
- two-dimensional pressure isotherm.
Addiction  from S M:
from S M:

At  - two-dimensional pressure increases sharply. At
- two-dimensional pressure increases sharply. At  two-dimensional is deformed, causing sudden growth
two-dimensional is deformed, causing sudden growth  .
.
A film bounded by identical phases on both sides is called double-sided. In such films, constant movement of the mother liquor is observed.
Films less than 5 nm thick are called black films.

Adsorption layers must have two characteristics: viscosity and easy mobility, fluidity and elasticity.
The Marangoni effect is self-healing.
Gibbs triangle,  - overpressure.
- overpressure.
The film has stretched and due to the fact that part of the liquid has left, the surfactants rush into the free space. Gibbs triangle.
The effect of adsorption strength of bodies.
There is always an adsorption layer on the surface of the film, for which then
Langmuir equation:


 into two-dimensional pressure
into two-dimensional pressure
 - an analogue of the Shishkovsky equation
- an analogue of the Shishkovsky equation
Electrokinetic phenomena. Electric double layer (EDL).
Gelemholtz model. Gouy-Chapman theory.
1808 Flight

U – shaped tube, immerse 2 electrodes into it. The law of communicating vessels is violated and a change in the level of liquid in the tube occurs - electrokinetic phenomena.
Kinetic phenomena:
Electrophoresis
Electroosmosis
Flow (flow) potential
Sedimentation potential
1 and 2 arise when a potential difference is applied; 3 and 4, punching and sedimentation of colloidal particles cause the appearance of a potential difference.
Electroosmosis is the movement of a dispersion medium relative to a stationary dispersed phase under the influence of an electric current.
Electrophoresis – this is the movement of dispersed phase particles relative to a stationary dispersion medium under the influence of an electric current.
P  The reason for the occurrence of electrokinetic phenomena is the spatial separation of charges and the appearance of a double electrical layer.
The reason for the occurrence of electrokinetic phenomena is the spatial separation of charges and the appearance of a double electrical layer.
The electrical double layer is a flat capacitor, one plate is formed by potential-determining ions, the other by counter-ions. The ions are contaminated in the same way that potential-determining co-ions are pushed into the volume of the solution. Distance between plates  . The potential drops linearly, the potential difference
. The potential drops linearly, the potential difference  .
.
An external potential difference causes the appearance of a shear modulus  is a pair of forces per unit area acting along the surface of a solid body.
is a pair of forces per unit area acting along the surface of a solid body.

In equilibrium, the shear modulus is equal to the viscous friction modulus (  ).
).

In our conditions  ,
,

 - Gelemholtz-Smalukowski equation
- Gelemholtz-Smalukowski equation
 - linear speed of phase displacement.
- linear speed of phase displacement.
E is the electric field strength.
 - potential difference between plates
- potential difference between plates
 - electrophoretic mobility [m 2 /(V*s)].
- electrophoretic mobility [m 2 /(V*s)].
The Helemholtz model does not take into account the thermal motion of molecules. In reality, the distribution of ions in the double layer is more complex.
Gui and Chapman identified the following causes of DES:
The transition of an ion from one phase to another when equilibrium is established.
Ionization of solid phase matter.
Completion of the surface with ions present in the dispersion medium.
Polarization from an external current source.
The electrical double layer has a fuzzy or diffuse structure. The ions tend to be evenly distributed throughout the diffuse layer.
The diffuse layer consists of counterinons; the length of the layer is determined by their kinetic energy. At temperatures approaching absolute zero, counter-ions are as close as possible to potential-determining ions.
Danya's theory is based on two equations:
Boltzmann equation

 - work against the forces of electrostatic interaction.
- work against the forces of electrostatic interaction.
 - volumetric charge density.
- volumetric charge density.

Poisson's equation 
Since the thickness of the EDL is much smaller than the particle size and for a flat EDL the derivative with respect to coordinates  And
And  is abolished.
is abolished. 
For e y at y<<1 функцию можно разложить в ряд Маклорена:

Let us limit ourselves to two terms of the series, then:


 - DEL thickness is the distance at which the DEL potential decreases in e once.
- DEL thickness is the distance at which the DEL potential decreases in e once.
The lower the temperature, the less  . At T→0 – flat DEL. The higher the concentration, the more I, the less
. At T→0 – flat DEL. The higher the concentration, the more I, the less  .
.





 “–” means that the potential decreases with distance. =>
“–” means that the potential decreases with distance. =>
=>

 ,
,
 - the potential decreases exponentially.
- the potential decreases exponentially.
Potential for surface charge density: 
Surface charge is a volume charge with the opposite sign, integrated over distance.




=>


Where the potential decreases by 2.7 times - 
Double layer capacity 
The disadvantage of the theory is that the presence of the Helemholtz layer is not taken into account, i.e. does not take into account  , hence the errors in determining the main parameters. It also does not explain the influence of ions of different nature on the thickness of the electrical double layer.
, hence the errors in determining the main parameters. It also does not explain the influence of ions of different nature on the thickness of the electrical double layer.
Stern's theory. Structure of a colloidal micelle.
The electrical double layer consists of two parts: dense and diffuse. A dense layer is formed as a result of the interaction of potential-forming ions with specifically adsorbed ones. These ions, as a rule, are partially or completely dehydrated and can have either the same or opposite charge to the potential-determining ions. It depends on the ratio of electrostatic interaction energy  and specific adsorption potential
and specific adsorption potential  . The dense layer ions are fixed. The other part of the ions is located in the diffuse layer; these ions are free and can move deeper into the solution, i.e. from an area of higher concentration to an area of lower concentration. The total charge density consists of two parts.
. The dense layer ions are fixed. The other part of the ions is located in the diffuse layer; these ions are free and can move deeper into the solution, i.e. from an area of higher concentration to an area of lower concentration. The total charge density consists of two parts.

 -charge of the Helmholtz layer
-charge of the Helmholtz layer
 -Diffuse layer charge
-Diffuse layer charge
The surface has a certain number of adsorption centers, each of which interacts with one counterion. The constant of such a quasi-chemical reaction is equal to:

 , Where
, Where  - mole fraction of counterions in solution
- mole fraction of counterions in solution
Helmholtz distribution
The potential decreases linearly
Gouy potential distribution. There is no dense layer, the potential decreases exponentially from the value 
Stern distribution.
Initially, the potential decrease is linear and then exponential.
When an electric field is applied in the case of electrophoresis, it is not the particle of the solid phase that moves directly, but the particle of the solid phase with a layer of ions surrounding it. The DES repeats the shape of the dispersed phase particle. When a potential is applied, part of the diffuse layer is torn off. The break line is called sliding boundary.
The potential that arises at the slip boundary as a result of separation of part of the diffuse layer is called electrokinetic potential(Zeta potential  ).
).
A dispersed phase particle with a surrounding layer of counterions and a double electrical layer is called micelle.
Rules for writing colloidal micelles:


1-1 charging electrolyte
T – dispersed phase particle.
AA is the boundary between the dense and diffuse parts.
BB – sliding boundary.
The sliding boundary may or may not coincide with line AA.
The pH value at which the zeta potential is zero is called isoelectric point.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. Excess CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - micelle notation.
CaSO 4 m – aggregate.
CaSO 4 m∙nCa 2+ – core.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - particle.
2. Excess Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micelle
CaSO 4 m – aggregate.
CaSO 4 m∙nSO 4 2 + – core.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - particle
Gelemholtz-Smoluchowski equation

 - linear speed of boundary displacement (in electroosmosis).
- linear speed of boundary displacement (in electroosmosis).
 - potential difference across the capacitor plates (in electroosmosis).
- potential difference across the capacitor plates (in electroosmosis).


 - volumetric flow rate of the solution, S– cross-sectional area of the cell.
- volumetric flow rate of the solution, S– cross-sectional area of the cell.

E is the electric field strength.


 (for electroosmosis).
(for electroosmosis).
For flow potential:

 - potential
- potential
 - pressure on the membrane
- pressure on the membrane
As a rule, the values of electrophoretic mobilities and electroosmotic mobilities are less than calculated ones. This happens due to:
Relaxation effect (when a dispersed phase particle moves, the symmetry of the ionic atmosphere is broken).
Electrophoretic inhibition (the occurrence of additional friction as a result of the movement of counterions).
Distortion of current lines in the case of electrically conductive particles.
Relationship between surface tension and potential. Lippmann equation.
The formation of EDL occurs spontaneously due to the desire of the system to reduce its surface energy. In conditions of constant T And p the generalized equation of the first and second laws of thermodynamics looks like:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1st Lippmann equation.
- 1st Lippmann equation.
 - surface charge density.
- surface charge density.
 - differential capacitance.
- differential capacitance.
 - 2nd Lippmann equation.
- 2nd Lippmann equation.
WITH– capacity.
Let's solve the 1st Lippmann equation and the fundamental adsorption equation:
 ,
,


 , Then
, Then
 - Nernst equation
- Nernst equation
 ,
,
 ,
,




 - equation of the electrocapillary curve (ECC).
- equation of the electrocapillary curve (ECC).
IN  :
: , But
, But 
Cationic surfactants (CPAS) reduce the cathodic branch of the EKC.
Anionic surfactants (APS) reduce the anodic branch of the EKC.
Nonionic surfactants (NSAS) reduce the middle part of the ECC.
Stability of dispersed systems. Disjoining pressure.
Dispersed systems can be divided:
Systems that are thermodynamically unstable can be kinetically stable due to the transition to a metastable state.
There are two types of stability:
Sedimentation stability (with respect to gravity).
Aggregative stability. (relative to adhesion)
Coagulation is the process of particles sticking together, leading to the loss of aggregative stability. Coagulation can be caused by temperature changes, pH, stirring, and ultrasound.
Coagulation is distinguished:
Reversible.
Irreversible.
Coagulation occurs with the introduction of electrolytes.
Coagulation rules:

Film- this is the part of the system located between two interfacial surfaces.
Disjoining pressure occurs when the film thickness sharply decreases as a result of the interaction of approaching surface layers.

“-” - as the film thickness decreases, the disjoining pressure increases.
P 0 is the pressure in the bulk phase, which is a continuation of the interlayer.
P 1 – pressure in the film.
Theory of stability. DLFO (Deryagin, Landau, Fairway, Overbeck).
According to the DLFO theory, disjoining pressure has two components:
Electrostatic P E (positive, it is due to the forces of electrostatic repulsion). Corresponds to a decrease in the Gibbs energy with increasing film thickness.
Molecular P M (negative, due to the action of attractive forces). It is caused by film compression due to chemical surface forces, the radius of action of forces is tenths of nm with an energy of about 400 kJ/mol.
Total interaction energy:

 - the system is aggregatively stable
- the system is aggregatively stable
 - unstable system
- unstable system
P  positive component.
positive component.
The increase is due to an increase in potential energy when thin films are compressed. For films of large thickness, the excess ion energy is compensated and is equal to the energy interaction in the volume of the dispersion medium.
If  (
( - film thickness,
- film thickness,  - ion radius) thinning of the film leads to the disappearance and reduction of molecules and ions with minimal surface energy in it. The number of neighboring particles decreases, as a result of which the potential energy of the particles remaining in the film increases.
- ion radius) thinning of the film leads to the disappearance and reduction of molecules and ions with minimal surface energy in it. The number of neighboring particles decreases, as a result of which the potential energy of the particles remaining in the film increases.


The DLVO theory considers the interaction of particles as the interaction of plates.

Particles don't interact



 - Laplace equation,
- Laplace equation,  ,
,


For weakly charged surfaces
For highly charged surfaces:

The molecular component is the interaction of two atoms:
 ~
~
Interaction of an atom with a surface:

Let's take two records:
D  To obtain the molecular component, it is necessary to sum up all the interaction energies of the atoms of the right and left plates.
To obtain the molecular component, it is necessary to sum up all the interaction energies of the atoms of the right and left plates.
Where  - Hamaker constant (takes into account the nature of interacting bodies).
- Hamaker constant (takes into account the nature of interacting bodies).
That. the interaction energy of particles in a system can be expressed using potential curves.
I – primary potential minimum. This is a zone of irreversible coagulation, the forces of attraction prevail.
II – zone of aggregative stability, repulsive forces predominate.
III – secondary potential minimum (or flocculation zone). There is an electrolyte layer between the particles of the dispersed phase, and the particles can be separated and transferred to the zone of aggregation stability.

Curve 1 – the system is aggregatively stable.
Curve 2 – stable in zone I, unstable in zone II.
Curve 3 – coagulation has occurred in the system.
Curve 4 – at point 4 the total interaction energy U=0,  , this extremum point corresponds to the beginning of rapid coagulation.
, this extremum point corresponds to the beginning of rapid coagulation.
There are two cases:
1. Slightly charged surfaces:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - this is the thickness of the layer corresponding to the beginning of the coagulation process.
- this is the thickness of the layer corresponding to the beginning of the coagulation process.






 - for weakly charged surfaces
- for weakly charged surfaces
 Then
Then 
2. For highly charged surfaces:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Let's square (3)

Coagulation:
In specific adsorption, ions can be adsorbed in super-equivalent amounts such that the surface can change its charge. The surface is recharged.
In the case of specific adsorption, ions not only of opposite signs can be adsorbed, but also of the same sign.
If ions of the same sign as the surface are adsorbed, then in the surface layer there will be not a drop in the potential, but an increase in it.

Neutralization coagulation (occurs with the participation of weakly charged particles and depends not only on the charge of the electrolyte-coagulator, but also on the potential at the boundary of the dense and diffuse layers).
Smoluchowski's theory of rapid coagulation.

Dependence of coagulation rate on electrolyte concentration.
I – coagulation rate is low,
II – the coagulation rate is almost proportional to the electrolyte concentration.
III – region of rapid coagulation, the speed is practically independent of concentration.
Basic provisions:
The initial sol is monodisperse, similar particles have a spherical shape.
All particle collisions are effective.
When two primary particles collide, a secondary particle is formed. Secondary + primary = tertiary. Primary, secondary, tertiary – multiplicity.
In terms of chemical kinetics, the coagulation process can be described by the equation:

The solution will be the equation: 
 - half coagulation time. This is the time during which the number of sol particles decreases by 2 times.
- half coagulation time. This is the time during which the number of sol particles decreases by 2 times.

 ,
,
 ,
,
 ,
,


As the multiplicity increases, the maximum of the coagulation curves shifts towards larger values  .
.
Flaws:
Assumption of monodispersity.
Assumption about the effectiveness of all collisions.
Interaction of polymers with liquids and gases
The processes of interaction of polymers with low molecular weight liquids play an important role in the processes of formation of finished products (for example, fibers from a solution), modification of the properties (plasticization) of the material, as well as in the operating conditions of these products in various liquid environments. The interaction is expressed in the absorption of liquid by the polymer and is called sorption. If sorption occurs in the volume of a polymer material, it is called absorption. If absorption occurs in surface layers, then the process is called adsorption.
Sorption
The mechanism of adsorption is due to the presence of surface tension forces at the interfaces between media (Fig. 5.1) due to the difference in the forces of intermolecular interaction in them. This leads to the accumulation of excess energy on the surface of a substance that tends to draw in its surface molecules (molecules adsorbent) and weaker interacting molecules (molecules adsorptive) inside the volume. The amount of adsorption largely depends on the specific surface area of the adsorbent. Numerically, adsorption is expressed by the number of moles of adsorbed substance per unit mass of the adsorbent - x/m.
The study of sorption makes it possible to obtain valuable information about the structure of a polymer and the degree of packing of its molecules.
Usually, sorption processes are described using curves of the dependence of the amount of adsorbed substance on its concentration (or pressure) in the gas phase at a constant temperature (sorption isotherms, Fig. 5.2.). Here the value R/R s is the ratio of the vapor pressure of the adsorbent to the elasticity of its saturated vapor at a given temperature.

In the region of low vapor pressures, Henry's linear law is satisfied:
Where A- amount of adsorbed substance; a m- limiting adsorption, proportional to the active surface of the adsorbent; p- sorbate pressure; k- adsorption constant. On fig. 5.2 the completion of monomolecular adsorption is determined by the exit of the sorption isotherm to the shelf in the range of relative pressures 0.4 ÷ 0.5.
In the presence of polymolecular adsorption and condensation on the surface of a porous adsorbent ( R/R s > 0.6 in Fig. 5.2) use the universal equation
 | (5.3) |
Thermodynamics of the adsorption process
Since, as a rule, the intermolecular interaction of adsorbent molecules is less intense than that of the adsorbent, adsorption proceeds with a decrease in the free energy of the surface (Δ F < 0) и выделением тепла (уменьшением энтальпии ΔN < 0). При равновесии процессов адсорбции и десорбции ΔF= 0. The value calculated in the course of adsorption characterizes the number and activity of groups on the surface of the adsorbent capable of reacting with the absorbent. During adsorption, the entropy of the system also decreases (Δ S < 0), поскольку молекулы абсорбтива ограничивают подвижность молекул полимера, уменьшая возможное число конформаций: ΔS = k ln( W 2 / W 1), where is Boltzmann’s constant, W 2 and W 1 - thermodynamic probability of the final and initial state of the system.
Adsorption takes place at the interface. Therefore, it is expedient to consider the thermodynamic description of surface phenomena as a particular case of the thermodynamics of heterogeneous systems.
Rice. 3.4. Gibbs adsorption: 1- two-phase reference system, 2- real two-phase system with inhomogeneous region
In the thermodynamics of heterogeneous systems it is used additivity principle which is as follows: all the extensive properties of a heterogeneous system are equal to the sum of the corresponding extensive properties that the phases would have had before they were brought into contact. Let us denote the phases by α and β (Fig. 4). Then for an ideal system, such that the properties of the phases near the interface coincide with their bulk properties, for the internal energy U, volume V, mass (number of moles) n, entropy S after equilibrium is established in a heterogeneous system, the relations are valid:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
This assumes that the temperature and pressure in both phases are the same.
For real heterogeneous systems, the transition region at the interface between two phases makes an additional contribution to the extensive properties of the system. If surface phenomena occur, one should take into account the difference between the extensive properties of a real heterogeneous system and the extensive properties of a model system in which there are no surface phenomena. Such a system is called a comparison system. The comparison system has the same intensive parameters (T, P, C i …) and the same volume V as the real system (Fig. 4).
From a thermodynamic point of view, the adsorption value G is understood as the excess amount of substance n s, expressed in moles or grams, which a real heterogeneous system has in comparison with the reference system, related to the surface area of the phase separation or to the surface area of the adsorbent A. It is assumed that the comparison system has the same intensive parameters (T, P, C i) and the same volume (V = V α + V β) as the real system (Fig. 4).
Г = (n - n α - n β)/A = n s /A 3.11
Excess thermodynamic functions of the transition region of a real system (we denote them by the index s) can be written as
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β etc.
Experimental measurements of adsorption always give adsorption precisely as an excess of the component in the real system compared to the chosen reference system. For example, when adsorption of gas on a solid adsorbent or when adsorption of components on a solid phase, to find adsorption values, determine the change in the initial concentrations of the adsorbate after the contact of phases α and β
n i s = V(C i o - C i),
Where C i o– initial concentration of the i-th component, C i– concentration of the i-th component after establishing equilibrium between the contacting phases. It is believed that the volume V does not change. However, the concentration i th component C i, obtained experimentally, is determined in volume V' above the interface without taking into account the volume of the inhomogeneous region of the transition layer V α at the interface where the concentration is C i α. Thus, due to the existence of an inhomogeneous region in a real system, the total volume of the system can be represented as V = V’ + V α. All quantity i-th component C i o will be distributed between these two volumes:
V C i o = V’ C i + V α C i α ,
and the number of moles of the component i, adsorbed on the interface, will be equal to
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Those. adsorption determined experimentally is an excess of the i-th component in the volume V α in comparison with the amount of this component in the same volume far from the interface. This type of adsorption is called Gibbs adsorption. .
V α C i α called complete content i- th component in the adsorption layer. In the region of very low concentrations C i in volume V' amendment V α C i equation (3.2) can be neglected and the measured value can be considered V α C i α full content i- th component in the adsorption layer, for example, in the adsorption of gas on a solid adsorbent at low pressures.
Kuznetsova E.S. and Buryak A.K. carried out a comparison of the thermodynamic characteristics of the adsorption of amino acids and their associates. The work investigated the influence of the structure of amino acids, their dimers and associates with eluent components on their adsorption on the surface of carbon materials. A molecular statistical calculation of the thermodynamic characteristics of adsorption (TCA) for aromatic amino acids (phenylalanine, tyrosine), heterocyclic amino acid (tryptophan) and their dimers with trifluoroacetic acid (TFA) on the surface of graphitized thermal carbon (GTS) was carried out. The data obtained are compared with the patterns of amino acid retention on porous graphitized carbon Hypercarb under reversed-phase high-performance liquid chromatography (RP HPLC) conditions. It has been shown that TCA and amino acid retention values increase with increasing carbon chain of these compounds.
Shkolin A.V. and Fomkin A.A. analyzed the behavior of thermodynamic functions (differential molar isosteric heat of adsorption, entropy, enthalpy and heat capacity) of the methane-microporous carbon adsorbent AUK adsorption system depending on the adsorption equilibrium parameters in the temperature range from 177.65 up to 393 K and pressures from 1 Pa to 6 MPa. Taking into account the influence of the nonideality of the gas phase and the non-inertness of the adsorbent led to the appearance of a temperature dependence of the isosteric heat of adsorption, especially in the region of high pressures of the adsorbent. For the system under study, the main influence on the thermodynamic functions of the adsorption system is exerted by the nonideality of the gas phase. The correction for the non-inertness of the adsorbent in this range of parameters of the adsorption system is no more than 2.5%.
At the Institute of General and Inorganic Chemistry of the Academy of Sciences of the Republic of Uzbekistan Muminov S.Z. in his work, he investigated changes in the surface properties and porous structure of montmorillonite when replacing the exchangeable cations of the mineral with polyhydroxyaluminum ones. Preliminary thermal vacuuming has a significant effect on the adsorption properties of polyhydroxyaluminum montmorillonite in relation to methyl alcohol. Based on data from a series of isosteres of CH3 adsorption on dehydrated sodium and modified montmorillonites, measured over a wide temperature range, the dependence of the heat of adsorption on the amount of adsorbed substance was established.
N.S. Kazbanov, A.V. Matveeva and O.K. Krasilnikov conducted a study of the adsorption of phenol from aqueous solutions by activated carbons such as FAS, PAH and carbon felt at temperatures of 293, 313 and 343 K in the concentration range 5 - 250 mmol/l. A series of samples of sequentially activated carbon FAS, characterized by a narrow pore size distribution, were obtained by carbonization of furfural-based polymers. PAH is a microporous polymeric activated carbon. Carbon felt is a fibrous material based on hydrated cellulose fibers. The parameters of the porous structure of the adsorbents were determined from nitrogen vapor adsorption isotherms at 77 K (ASAP-2020, Micromeritics, USA). Studies of the adsorption of solutions were carried out using the ampoule method in a thermostat. Selected samples were analyzed by spectrophotomery. The analysis of the obtained liquid-phase adsorption isotherms was carried out using the theory of volumetric filling of micropores (VFM) according to the Dubinin-Radushkevich (DR) equation.
The effect of temperature on sorption from liquid solutions is ambiguous. On the one hand, for microporous adsorbents, the penetration of molecules into pores comparable in size to these molecules depends on kinetic energy and, accordingly, increases with temperature. On the other hand, physical adsorption is an exothermic process and adsorption decreases with temperature. The relationship between these factors for each system determines the course of the temperature dependence of adsorption.
The uniqueness of the adsorbent - phenol system is that it has an inverse temperature dependence of adsorption isotherms because As the temperature increases from 293 to 313 K, the limiting value of adsorption increases, which is apparently due to the molecular sieve effect: with increasing temperature, phenol molecules are able to penetrate into narrower pores of carbon materials. Adsorption occurs mainly in micropores, since adsorbents have a small number of mesopores. As the micropore size increases, the maximum adsorption values increase significantly, reaching 2.9 mmol/g for PAH, 8.5 mmol/g for FAS, and 12.7 mmol/g for felt. The resulting adsorption isotherms are well described by the DD equation with the exponent equal to 2.


