Alkenet janë kimikisht aktive. Vetitë e tyre kimike përcaktohen kryesisht nga prania e një lidhjeje të dyfishtë. Për alkenet, reaksionet e shtimit elektrofil dhe reaksionet e shtimit radikal janë më karakteristikë. Reaksionet e shtimit nukleofilik zakonisht kërkojnë një nukleofile të fortë dhe nuk janë tipike për alkenet. Alkenet hyjnë lehtësisht në reaksione oksidimi, shtimi dhe janë gjithashtu të afta për zëvendësimin e radikaleve alilike.
Reaksionet e shtimit
Hidrogjenimi Shtimi i hidrogjenit (reaksioni i hidrogjenizimit) tek alkenet kryhet në prani të katalizatorëve. Më shpesh, përdoren metale të grimcuara - platini, nikeli, paladiumi etj. Si rezultat, formohen alkanet përkatëse (hidrokarburet e ngopura).
$CH_2=CH_2 + H2 → CH_3–CH_3$
shtimi i halogjeneve. Alkenet reagojnë lehtësisht me klorin dhe bromin në kushte normale për të formuar dihaloalkanet përkatëse, në të cilat atomet e halogjenit ndodhen në atomet fqinje të karbonit.
Vërejtje 1
Kur alkenet ndërveprojnë me bromin, ngjyra e verdhë-kafe e bromit zbardhet. Ky është një nga reaksionet cilësore më të vjetra dhe më të thjeshta për hidrokarburet e pangopura, pasi alkinet dhe alkadienet gjithashtu reagojnë në mënyrë të ngjashme.
$CH_2=CH_2 + Br_2 → CH_2Br–CH_2Br$
shtimi i halogjeneve të hidrogjenit. Kur hidrokarburet e etilenit reagojnë me halogjenët e hidrogjenit ($HCl$, $HBr$), formohen haloalkanet, drejtimi i reaksionit varet nga struktura e alkeneve.
Në rastin e etilenit ose alkeneve simetrike, reaksioni i shtimit ndodh pa mëdyshje dhe çon në formimin e vetëm një produkti:
$CH_2=CH_2 + HBr → CH_3–CH_2Br$
Në rastin e alkeneve josimetrike, formimi i dy produkteve të ndryshme të reaksionit të shtimit është i mundur:
Vërejtje 2
Në fakt, në thelb formohet vetëm një produkt reagimi. Rregullsia e drejtimit të kalimit të reaksioneve të tilla u vendos nga kimisti rus V.V. Markovnikov në 1869 Quhet sundimi i Markovnikov. Në bashkëveprimin e halogjeneve të hidrogjenit me alkenet josimetrike, atomi i hidrogjenit bashkohet në vendin ku prishet lidhja e dyfishtë në atomin më të hidrogjenizuar të karbonit, domethënë para se të lidhet me një numër të madh atomesh hidrogjeni.
Markovnikov e formuloi këtë rregull në bazë të të dhënave eksperimentale, dhe vetëm shumë më vonë ai mori një justifikim teorik. Merrni parasysh reagimin e propilenit me klorur hidrogjeni.
Një nga veçoritë e lidhjes $p$ është aftësia e saj për t'u polarizuar lehtësisht. Nën ndikimin e grupit metil (efekti induktiv pozitiv + $I$) në molekulën e propenit, densiteti elektronik i lidhjes $p$ zhvendoset në një nga atomet e karbonit (= $CH_2$). Si rezultat, një ngarkesë e pjesshme negative ($\delta -$) shfaqet në të. Në atomin tjetër të karbonit të lidhjes dyfishe, lind një ngarkesë e pjesshme pozitive ($\delta +$).
Kjo shpërndarje e densitetit të elektronit në molekulën e propilenit përcakton vendndodhjen e sulmit të ardhshëm nga protoni. Ky është atomi i karbonit i grupit të metilenit (= $CH_2$), i cili mbart një ngarkesë të pjesshme negative $\delta-$. Dhe klori, në përputhje me rrethanat, sulmon atomin e karbonit me një ngarkesë të pjesshme pozitive $\delta+$.
Si pasojë, produkti kryesor i reagimit të propilenit me klorur hidrogjeni është 2-kloropropani.
Hidratimi
Hidratimi i alkeneve ndodh në prani të acideve minerale dhe i bindet rregullit Markovnikov. Produktet e reagimit janë alkoolet
$CH_2=CH_2 + H_2O → CH_3–CH_2–OH$
Alkilimi
Shtimi i alkaneve tek alkenet në prani të një katalizatori acid ($HF$ ose $H_2SO_4$) në temperatura të ulëta çon në formimin e hidrokarbureve me një peshë molekulare më të lartë dhe shpesh përdoret në industri për të prodhuar lëndë djegëse motorike.
$R–CH_2=CH_2 + R’–H → R–CH_2–CH_2–R’$
Reaksionet e oksidimit
Oksidimi i alkeneve mund të ndodhë, në varësi të kushteve dhe llojeve të reagentëve oksidues, si me thyerjen e lidhjes së dyfishtë, ashtu edhe me ruajtjen e skeletit të karbonit:
reaksionet e polimerizimit
Molekulat e alkenit janë të afta të shtohen me njëra-tjetrën në kushte të caktuara me hapjen e lidhjeve $\pi$ dhe formimin e dimerëve, trimerëve ose komponimeve me molekulare të lartë - polimere. Polimerizimi i alkeneve mund të kryhet si nga mekanizmat e radikalëve të lirë ashtu edhe nga mekanizmat kation-anion. Si iniciatorë të polimerizimit përdoren acidet, peroksidet, metalet etj.. Edhe reaksioni i polimerizimit kryhet nën ndikimin e temperaturës, rrezatimit dhe presionit. Një shembull tipik është polimerizimi i etilenit për të formuar polietileni
$nCH_2=CH_2 → (–CH_2–CH_(2^–))_n$
Reaksionet e zëvendësimit
Reaksionet e zëvendësimit për alkenet nuk janë tipike. Megjithatë, në temperatura të larta (mbi 400 °C), reaksionet radikale të shtimit, të cilat janë të kthyeshme, shtypen. Në këtë rast, bëhet e mundur të kryhet zëvendësimi i atomit të hidrogjenit në pozicionin alilik duke ruajtur lidhjen e dyfishtë.
$CH_2=CH–CH_3 + Cl_2 – CH_2=CH–CH_2Cl + HCl$
Hipermarketi i njohurive >>Kimi >>Kimia Klasa 10 >> Kimi: Alkenet
Hidrokarburet e pangopura përfshijnë hidrokarbure që përmbajnë lidhje të shumta midis atomeve të karbonit në molekula. Të pangopura janë alkenet, alkinet, alkadienet (polyenet). Hidrokarburet ciklike që përmbajnë një lidhje të dyfishtë në cikël (cikloalkenet), si dhe cikloalkanet me një numër të vogël atomesh karboni në cikël (tre ose katër atome) gjithashtu kanë karakter të pangopur. Vetia e "unsaturimit" lidhet me aftësinë e këtyre substancave për të hyrë në reaksione shtesë, kryesisht hidrogjen, me formimin e hidrokarbureve të ngopura, ose të ngopura - alkaneve.
Struktura
Alkenet - aciklike, që përmbajnë në molekulë, përveç lidhjeve të vetme, një lidhje të dyfishtë midis atomeve të karbonit dhe që korrespondon me formulën e përgjithshme C n H 2n.
Alkenet morën emrin e tyre të dytë - "olefinat" për analogji me acide yndyrore të pangopura (oleik, linoleik), mbetjet e të cilave janë pjesë e yndyrave të lëngshme - vajrave (nga anglishtja vaj - vaj).
Atomet e karbonit midis të cilëve ekziston një lidhje e dyfishtë, siç e dini, janë në një gjendje hibridizimi sp 2. Kjo do të thotë se një s- dhe dy p-orbitale marrin pjesë në hibridizim, ndërsa një orbitale p mbetet e pahibridizuar. Mbivendosja e orbitaleve hibride çon në formimin e një lidhje α, dhe për shkak të orbitaleve α të pahibridizuara të molekulave fqinje të etilenit, atomet e karbonit formojnë një të dytë, P-lidhje. Kështu, një lidhje dyfishe përbëhet nga një lidhje z dhe një lidhje p.
Orbitalet hibride të atomeve që formojnë lidhjen dyfishe janë në të njëjtin rrafsh, ndërsa orbitalet që formojnë lidhjen n janë pingul me rrafshin e molekulës (shih Fig. 5).
Një lidhje e dyfishtë (0,132 nm) është më e shkurtër se një lidhje e vetme dhe energjia e saj është më e madhe, domethënë është më e qëndrueshme. Sidoqoftë, prania e një lidhjeje 7r të lëvizshme, lehtësisht të polarizueshme çon në faktin se alkenet janë kimikisht më aktivë se alkanet dhe janë në gjendje të hyjnë në reaksione shtesë.
Seritë homologe të etenit
Alkenet e padegëzuara përbëjnë serinë homologe të etenit (etilen).
C2H4 - eten, C3H6 - propen, C4H8 - buten, C5H10 - penten, C6H12 - heksen etj.
Izomerizmi dhe nomenklatura
Për alkenet, si dhe për alkanet, izomeria strukturore është karakteristike. Izomerët strukturorë, siç e mbani mend, ndryshojnë nga njëri-tjetri në strukturën e skeletit të karbonit. Alkeni më i thjeshtë, i cili karakterizohet nga izomerë strukturorë, është buteni.
CH3-CH2-CH=CH2 CH3-C=CH2
l
CH3
buten-1 metilpropen
Një lloj i veçantë i izomerizmit strukturor është izomeria e pozicionit të lidhjes së dyfishtë:
CH3-CH2-CH=CH2 CH3-CH=CH-CH3
buten-1 buten-2
Rrotullimi pothuajse i lirë i atomeve të karbonit është i mundur rreth një lidhjeje të vetme karbon-karbon, kështu që molekulat e alkanit mund të marrin një larmi formash. Rrotullimi rreth lidhjes së dyfishtë është i pamundur, gjë që çon në shfaqjen e një lloji tjetër të izomerizmit në alkenet - gjeometrik, ose izomerizmi cis-trans.
Cis-izomerët ndryshojnë nga izomerët toraks nga rregullimi hapësinor i fragmenteve molekulare (në këtë rast, grupet metil) në lidhje me rrafshin P marrëdhëniet, dhe rrjedhimisht vetitë.
Alkenet janë izomere ndaj cikloalkaneve (izomeria ndërklasore), për shembull:
ch2=ch-ch2-ch2-ch2-ch3
heksen-1 cikloheksan
Nomenklatura alkenet, i zhvilluar nga IUPAC, është i ngjashëm me nomenklaturën e alkaneve.
1. Zgjedhja e qarkut kryesor
Formimi i emrit të një hidrokarburi fillon me përcaktimin e zinxhirit kryesor - zinxhiri më i gjatë i atomeve të karbonit në një molekulë. Në rastin e alkeneve, zinxhiri kryesor duhet të përmbajë një lidhje të dyfishtë.
2. Numërimi i atomeve të vargut kryesor
Numërimi i atomeve të vargut kryesor fillon nga fundi me të cilin është më afër lidhja dyfishe. Për shembull, emri i saktë i lidhjes është
ch3-chn-ch2-ch=ch-ch3 ch3
5-metilheksen-2, jo 2-metilheksen-4 siç mund të pritej.
Nëse është e pamundur të përcaktohet fillimi i numërimit të atomeve në zinxhir nga vendndodhja e lidhjes së dyfishtë, atëherë përcaktohet nga pozicioni i zëvendësuesve në të njëjtën mënyrë si për hidrokarburet e ngopura.
CH3-CH2-CH=CH-CH-CH3
l
CH3
2-metilheksen-3
3. Formimi i emrit
Emrat e alkeneve formohen në të njëjtën mënyrë si emrat e alkaneve. Në fund të emrit, tregohet numri i atomit të karbonit në të cilin fillon lidhja e dyfishtë dhe prapashtesa, që tregon se përbërja i përket klasës së alkeneve, -ene.
Faturë
1. Plasaritja e produkteve të naftës. Në procesin e plasaritjes termike të hidrokarbureve të ngopura, së bashku me formimin e alkaneve, ndodh formimi i alkeneve.
2. Dehidrogjenimi i hidrokarbureve të ngopura. Kur alkanet kalohen mbi një katalizator në një temperaturë të lartë (400-600 °C), një molekulë hidrogjeni ndahet dhe formohet një alken: 
3. Dehidratimi i alkooleve (ndarja e ujit). Efekti i agjentëve largues të ujit (H2804, Al203) në alkoolet monohidrike në temperatura të larta çon në eliminimin e një molekule uji dhe formimin e një lidhjeje të dyfishtë:
Ky reaksion quhet dehidrim intramolekular (në ndryshim nga dehidratimi ndërmolekular, i cili çon në formimin e etereve dhe do të studiohet në § 16 "Alkoolet").
4. Dehidrohalogjenimi (eliminimi i halogjenit të hidrogjenit).
Kur një haloalkan reagon me një alkali në një tretësirë alkoolike, formohet një lidhje e dyfishtë si rezultat i eliminimit të një molekule halidi hidrogjeni.
Vini re se ky reagim prodhon kryesisht buten-2 në vend të buten-1, që korrespondon me Rregulli i Zaitsev:
Kur një halogjen hidrogjeni ndahet nga haloalkanet dytësore dhe terciare, një atom hidrogjeni ndahet nga atomi i karbonit më pak të hidrogjenizuar.
5. Dehalogjenimi. Nën veprimin e zinkut në një derivat dibromo të një alkani, atomet e halogjenit ndahen nga atomet fqinje të karbonit dhe formohet një lidhje e dyfishtë: 
Vetitë fizike
Tre përfaqësuesit e parë të serisë homologe të alkeneve janë gaze, substancat e përbërjes C5H10-C16H32 janë të lëngshme, alkenet më të larta janë të ngurta.
Pikat e vlimit dhe shkrirjes rriten natyrshëm me një rritje të peshës molekulare të përbërjeve.
Vetitë kimike
Reaksionet e shtimit
Kujtojmë se një tipar dallues i përfaqësuesve të hidrokarbureve të pangopura - alkeneve është aftësia për të hyrë në reaksione shtesë. Shumica e këtyre reaksioneve zhvillohen me mekanizmin e shtimit elektrofil.
1. Hidrogjenizimi i alkeneve. Alkenet janë në gjendje të shtojnë hidrogjen në prani të katalizatorëve të hidrogjenizimit - metaleve - platinit, paladiumit, nikelit:
CH3-CH2-CH=CH2 + H2 -> CH3-CH2-CH2-CH3
Ky reagim vazhdon si në presion atmosferik ashtu edhe në presion të ngritur dhe nuk kërkon temperaturë të lartë, pasi është ekzotermik. Me një rritje të temperaturës në të njëjtët katalizatorë, mund të ndodhë reaksioni i kundërt, dehidrogjenizimi.
2. Halogjenimi (shtimi i halogjeneve). Ndërveprimi i një alkeni me ujin me brom ose një tretësirë bromi në një tretës organik (СCl4) çon në një zbardhje të shpejtë të këtyre tretësirave si rezultat i shtimit të një molekule halogjene në alken dhe formimit të dihaloalkaneve.
Markovnikov Vladimir Vasilievich
(1837-1904)
Kimisti organik rus. Rregulla të formuluara (1869) për drejtimin e reaksioneve të zëvendësimit, eliminimit, shtimit të lidhjes së dyfishtë dhe izomerizimit, në varësi të strukturës kimike. Hulumtoi (që nga viti 1880) përbërjen e naftës, hodhi themelet e petrokimisë si shkencë e pavarur. U hap (1883) një klasë e re e substancave organike - ciklo-parafinat (naftenet).
3. Hidrohalogjenimi (shtimi i halogjenit të hidrogjenit).
Reaksioni i shtimit të halogjenit të hidrogjenit do të diskutohet më në detaje më poshtë. Ky reagim i bindet rregullit të Markovnikov:
Kur një halogjen hidrogjeni i shtohet një alkeni, hidrogjeni lidhet me një atom karboni më të hidrogjenizuar, d.m.th., një atom në të cilin ka më shumë atome hidrogjeni dhe një halogjen me një atom më pak të hidrogjenizuar.
4. Hidratim (shtim uji). Hidratimi i alkeneve çon në formimin e alkooleve. Për shembull, shtimi i ujit në eten është themeli i një prej metodave industriale për prodhimin e alkoolit etilik:
CH2=CH2 + H2O -> CH3-CH2OH
etanol etanol
Vini re se një alkool primar (me një grup hidroksil në karbonin primar) formohet vetëm kur eteni hidratohet. Kur propeni ose alkenet e tjera hidratohen, formohen alkoole dytësore. 
Ky reagim gjithashtu vazhdon në përputhje me rregullin e Markovnikov - kationi i hidrogjenit i shtohet atomit të karbonit më të hidrogjenizuar, dhe grupi hidroksi i shtohet atij më pak të hidrogjenizuar.
5. Polimerizimi. Një rast i veçantë i shtimit është reaksioni i polimerizimit të alkeneve:
Ky reaksion shtimi zhvillohet nga një mekanizëm i radikaleve të lira.
Reaksionet e oksidimit
Ashtu si çdo përbërje organike, alkenet digjen në oksigjen për të formuar CO2 dhe H20.
Ndryshe nga alkanet, të cilat janë rezistente ndaj oksidimit në tretësirë, alkenet oksidohen lehtësisht nga tretësirat ujore të permanganatit të kaliumit. Në tretësirat neutrale ose pak alkaline, alkenet oksidohen në diole (alkoolet dihidrike), dhe grupet hidroksil lidhen me ato atome midis të cilave ekzistonte një lidhje e dyfishtë përpara oksidimit.
Siç e dini tashmë, hidrokarburet e pangopura - alkenet janë në gjendje të hyjnë në reaksione shtesë. Shumica e këtyre reaksioneve zhvillohen me mekanizmin e shtimit elektrofil.
shtimi elektrofilik
Reaksionet elektrofile janë reaksione që ndodhin nën veprimin e elektrofileve - grimcave që kanë mungesë të densitetit të elektroneve, siç është një orbitale e pambushur. Grimca më e thjeshtë elektrofile është kationi i hidrogjenit. Dihet se atomi i hidrogjenit ka një elektron për 3-në-orbitale. Një kation hidrogjeni formohet kur një atom e humb atë elektron, kështu që kationi i hidrogjenit nuk ka fare elektrone:
H - 1e - -> H +
Në këtë rast, kationi ka një afinitet mjaft të lartë elektronik. Kombinimi i këtyre faktorëve e bën kationin e hidrogjenit një grimcë mjaft të fortë elektrofile.
Formimi i një kationi të hidrogjenit është i mundur gjatë shpërbërjes elektrolitike të acideve:
HBr -> H + + Br -
Është për këtë arsye që shumë reaksione elektrofile ndodhin në prani dhe me pjesëmarrjen e acideve.
Grimcat elektrofile, siç u përmend më herët, veprojnë në sisteme që përmbajnë rajone me densitet elektronik të rritur. Një shembull i një sistemi të tillë mund të jetë një lidhje e shumëfishtë (e dyfishtë ose e trefishtë) karbon-karbon.
Ju tashmë e dini se atomet e karbonit midis të cilëve formohet një lidhje dyfishe janë në një gjendje hibridizimi sp 2. P-orbitalet e pahibridizuara të atomeve fqinje të karbonit, të cilat janë në të njëjtin rrafsh, mbivendosen, duke formuar P-lidhja, e cila është më pak e fortë se lidhja z dhe, më e rëndësishmja, polarizohet lehtësisht nën veprimin e një fushe elektrike të jashtme. Kjo do të thotë se kur afrohet një grimcë e ngarkuar pozitivisht, elektronet e lidhjes TC zhvendosen në drejtimin e saj dhe të ashtuquajturat. P- komplekse.
Doli qe P-komplekse dhe pas shtimit të një kationi hidrogjeni në P-lidhjet. Kationi i hidrogjenit, si të thuash, pengohet në një densitet elektronik që del nga rrafshi i molekulës P-lidh dhe bashkohet me të. ![]()
Në fazën tjetër, ndodh zhvendosja e plotë e çiftit elektronik. P-lidhja me një nga atomet e karbonit, gjë që çon në shfaqjen e një çifti të vetëm elektronesh mbi të. Orbitalja e atomit të karbonit në të cilën ndodhet ky çift dhe orbitalja e pambushur e kationit të hidrogjenit mbivendosen, gjë që çon në formimin e një lidhje kovalente nga mekanizmi dhurues-pranues. Në të njëjtën kohë, atomi i dytë i karbonit mbetet një orbital i paplotësuar, d.m.th., një ngarkesë pozitive.
Grimca që rezulton quhet karbokacion sepse përmban një ngarkesë pozitive në atomin e karbonit. Kjo grimcë mund të kombinohet me çdo anion, një grimcë që ka një çift elektronik të pandarë, d.m.th., një nukleofile.
Konsideroni mekanizmin e reaksionit të shtimit elektrofilik duke përdorur shembullin e hidrobrominimit (shtimi i bromit të hidrogjenit) të etenit:
CH2= CH2 + HBr --> CHBr-CH3
Reaksioni fillon me formimin e një grimce elektrofile - një kation hidrogjeni, i cili ndodh si rezultat i shpërbërjes së një molekule bromidi hidrogjeni.
Sulmet e kationeve të hidrogjenit P-lidhje, formim P- një kompleks që shndërrohet shpejt në një karbokacion: 
Tani merrni parasysh një rast më të ndërlikuar.
Reaksioni i shtimit të bromurit të hidrogjenit në eten vazhdon në mënyrë të paqartë, dhe ndërveprimi i bromurit të hidrogjenit me propenin teorikisht mund të japë dy produkte: 1-bromopropan dhe 2-bromopropan. Të dhënat eksperimentale tregojnë se kryesisht përftohet 2-bromopropan.
Për ta shpjeguar këtë, do të duhet të marrim parasysh një grimcë të ndërmjetme - një karbokacion.
Shtimi i një kationi hidrogjeni në propen mund të çojë në formimin e dy karbokacioneve: nëse kationi i hidrogjenit është i lidhur me atomin e parë të karbonit, me atomin që është në fund të zinxhirit, atëherë i dyti, d.m.th. qendra e molekulës (1), do të ketë një ngarkesë pozitive; nëse bashkohet me të dytin, atëherë atomi i parë (2) do të ketë një ngarkesë pozitive. 
Drejtimi i preferuar i reaksionit do të varet nga cili karbokacion do të jetë më shumë në mjedisin e reagimit, i cili, nga ana tjetër, përcaktohet nga qëndrueshmëria e karbokacionit. Eksperimenti tregon formimin mbizotërues të 2-bromopropanit. Kjo do të thotë se formimi i karbokation (1) me një ngarkesë pozitive në atomin qendror ndodh në një masë më të madhe.
Stabiliteti më i madh i këtij karbokacioni shpjegohet me faktin se ngarkesa pozitive në atomin qendror të karbonit kompensohet nga efekti induktiv pozitiv i dy grupeve metil, efekti total i të cilave është më i lartë se efekti +/- i një grupi etil:
Modelet e reaksioneve të hidrohalogjenizimit të alkeneve u studiuan nga kimisti i famshëm rus V. V. Markovnikov, student i A. M. Butlerov, i cili, siç u përmend më lart, formuloi rregullin që mban emrin e tij.
Ky rregull u vendos në mënyrë empirike, domethënë empirike. Aktualisht, ne mund të japim një shpjegim plotësisht bindës për të.
Është interesante se reaksionet e tjera të shtimit elektrofil i binden gjithashtu rregullit të Markovnikovit, kështu që do të ishte e saktë të formulohej në një formë më të përgjithshme.
Në reaksionet e shtimit elektrofil, një elektrofil (një grimcë me një orbitale të pambushur) është ngjitur me një atom karboni më të hidrogjenizuar dhe një nukleofile (një grimcë me një palë të vetme elektronesh) është ngjitur me një atom më pak të hidrogjenizuar.
Polimerizimi
Një rast i veçantë i reaksionit të shtimit është polimerizimi i alkeneve dhe derivateve të tyre. Ky reagim zhvillohet me mekanizmin e shtimit të radikaleve të lira:
Polimerizimi kryhet në prani të iniciatorëve - komponimeve perokside, të cilat janë burim i radikalëve të lirë. Komponimet e peroksidit quhen substanca, molekulat e të cilave përfshijnë grupin -O-O-. Përbërja më e thjeshtë e peroksidit është peroksidi i hidrogjenit HOOH.
Në një temperaturë prej 100 ° C dhe një presion prej 100 MPa, ndodh homoliza e një lidhjeje të paqëndrueshme oksigjen-oksigjen dhe formimi i radikalëve - iniciatorëve të polimerizimit. Nën veprimin e radikaleve KO, fillon polimerizimi, i cili zhvillohet si reaksion i shtimit të radikalit të lirë. Rritja e zinxhirit ndalon kur përzierja e reaksionit rikombinohet nga radikalet - zinxhiri polimer dhe radikalët ose KOCH2CH2-.
Duke përdorur reaksionin e polimerizimit të radikaleve të lira të substancave që përmbajnë një lidhje të dyfishtë, fitohet një numër i madh i përbërjeve makromolekulare: 
Përdorimi i alkeneve me zëvendësues të ndryshëm bën të mundur sintetizimin e një game të gjerë materialesh polimerike me një gamë të gjerë vetive. 
Të gjitha këto komponime polimerike përdoren gjerësisht në fusha të ndryshme të veprimtarisë njerëzore - industri, mjekësi, përdoren për prodhimin e pajisjeve për laboratorët biokimikë, disa janë ndërmjetës për sintezën e komponimeve të tjera makromolekulare.
Oksidimi
Tashmë e dini se në tretësirat neutrale ose pak alkaline, alkenet oksidohen në diole (alkoolet dihidrike). Në një mjedis acid (një tretësirë e acidifikuar me acid sulfurik), lidhja e dyfishtë shkatërrohet plotësisht dhe atomet e karbonit midis të cilave ekzistonte lidhja e dyfishtë shndërrohen në atome karboni të grupit karboksil: 
Për të përcaktuar strukturën e tyre mund të përdoret oksidimi shkatërrues i alkeneve. Kështu, për shembull, nëse acidet acetike dhe propionike fitohen gjatë oksidimit të disa alkeneve, kjo do të thotë se penteni-2 ka pësuar oksidim, dhe nëse përftohet acid butirik (butanoik) dhe dioksid karboni, atëherë hidrokarburi fillestar është penteni. -1.
Aplikacion
Alkenet përdoren gjerësisht në industrinë kimike si lëndë e parë për prodhimin e substancave dhe materialeve të ndryshme organike.
Kështu, për shembull, eteni është materiali fillestar për prodhimin e etanolit, etilen glikolit, epooksideve, dikloroetanit.
Një sasi e madhe etenit përpunohet në polietileni, i cili përdoret për prodhimin e filmave të paketimit, enëve, tubave dhe materialeve izoluese elektrike.
Nga propeni përftohen glicerina, acetoni, izopropanoli, tretësit. Polimerizimi i propenit prodhon polipropilen, i cili është më i lartë se polietileni në shumë aspekte: ai ka një pikë shkrirjeje më të lartë dhe rezistencë kimike.
Aktualisht, fibrat me veti unike prodhohen nga polimere - analoge të polietilenit. Për shembull, fibra polipropileni është më e fortë se të gjitha fibrat sintetike të njohura.
Materialet e bëra nga këto fibra janë premtuese dhe përdoren gjithnjë e më shumë në fusha të ndryshme të veprimtarisë njerëzore.

1. Cilat lloje të izomerizmit janë karakteristikë për alkenet? Shkruani formulat për izomerët e mundshëm të pentenit-1.
2. Nga cilat komponime mund të përftohen: a) izobuteni (2-metilpropen); b) buten-2; c) buten-1? Shkruani ekuacionet për reaksionet përkatëse.
3. Deshifroni zinxhirin e mëposhtëm të shndërrimeve. Emërtoni përbërjet A, B, C. 4. Sugjeroni një metodë për marrjen e 2-kloropropanit nga 1-kloro-propani. Shkruani ekuacionet për reaksionet përkatëse.
5. Sugjeroni një metodë për pastrimin e etanit nga papastërtitë e etilenit. Shkruani ekuacionet për reaksionet përkatëse.
6. Jepni shembuj të reaksioneve që mund të përdoren për të dalluar hidrokarburet e ngopura dhe ato të pangopura.
7. Hidrogjenizimi i plotë i 2,8 g alkenit harxhuan 0,896 l hidrogjen (n.a.). Cila është pesha molekulare dhe formula strukturore e këtij përbërësi, i cili ka një zinxhir normal atomesh karboni?
8. Çfarë gazi ndodhet në cilindër (eteni ose propeni), nëse dihet se u deshën 90 cm3 (n.a.) oksigjen për të djegur plotësisht 20 cm3 nga ky gaz?
9*. Kur një alken reagon me klorin në errësirë, formohen 25,4 g diklorur dhe kur ky alken me të njëjtën masë reagon me bromin në tetraklorur karboni, formohen 43,2 g dibromid. Vendosni të gjitha formulat e mundshme strukturore të alkenit fillestar.
Historia e zbulimit
Nga materiali i mësipërm, ne kemi kuptuar tashmë se etilen është paraardhësi i serisë homologe të hidrokarbureve të pangopura, e cila ka një lidhje të dyfishtë. Formula e tyre është C n H 2n dhe quhen alkene.
Mjeku dhe kimisti gjerman Becher në vitin 1669 ishte i pari që mori etilenin nga veprimi i acidit sulfurik në alkoolin etilik. Becher zbuloi se etileni është më reaktiv se metani. Por, për fat të keq, në atë kohë, shkencëtari nuk mund të identifikonte gazin e marrë, prandaj ai nuk i caktoi asnjë emër.
Pak më vonë, kimistët holandezë gjithashtu përdorën të njëjtën metodë për marrjen e etilenit. Dhe meqenëse, kur bashkëvepronte me klorin, ai kishte aftësinë të formonte një lëng vajor, në përputhje me rrethanat mori emrin "gaz oksigjen". Më vonë u bë e ditur se ky lëng është dikloroetani.
Në frëngjisht, termi "vajor" tingëllon si oléfiant. Dhe pasi u zbuluan hidrokarbure të tjera të këtij lloji, Antoine Fourcroix, një kimist dhe shkencëtar francez, prezantoi një term të ri që u bë i zakonshëm për të gjithë klasën e olefinave ose alkeneve.
Por tashmë në fillim të shekullit të nëntëmbëdhjetë, kimisti francez J. Gay-Lussac tregoi se etanoli përbëhet jo vetëm nga gazi "vajor", por edhe nga uji. Përveç kësaj, i njëjti gaz u gjet në klorur etilik.
Dhe megjithëse kimistët përcaktuan se etilen përbëhet nga hidrogjen dhe karbon, dhe tashmë e dinin përbërjen e substancave, ata nuk mund të gjenin formulën e tij të vërtetë për një kohë të gjatë. Dhe vetëm në 1862, E. Erlenmeyer arriti të provojë praninë e një lidhje të dyfishtë në molekulën e etilenit. Kjo u njoh edhe nga shkencëtari rus A. M. Butlerov dhe konfirmoi saktësinë e këtij këndvështrimi eksperimentalisht.
Gjetja në natyrë dhe roli fiziologjik i alkeneve
Shumë njerëz janë të interesuar në pyetjen se ku mund të gjenden alkenet në natyrë. Pra, rezulton se ato praktikisht nuk ndodhin në natyrë, pasi përfaqësuesi i tij më i thjeshtë, etilen, është një hormon për bimët dhe sintetizohet në to vetëm në sasi të vogla.
Vërtetë, në natyrë ekziston një alken i tillë si muscalur. Ky një nga alkenet natyrale është një tërheqës seksual i mizës femërore.
Vlen t'i kushtohet vëmendje faktit se, duke pasur një përqendrim të lartë, alkenet e ulëta kanë një efekt narkotik që mund të shkaktojë konvulsione dhe acarim të mukozave.
Aplikimi i alkeneve
Jeta e shoqërisë moderne sot është e vështirë të imagjinohet pa përdorimin e materialeve polimerike. Meqenëse, ndryshe nga materialet natyrore, polimeret kanë veti të ndryshme, ato janë të lehta për t'u përpunuar dhe nëse shikoni çmimin, ato janë relativisht të lira. Një aspekt tjetër i rëndësishëm në favor të polimereve është se shumë prej tyre mund të riciklohen.
Alkenet kanë gjetur aplikimin e tyre në prodhimin e plastikës, gomave, filmave, teflonit, alkoolit etilik, acetaldehidit dhe komponimeve të tjera organike.

Në bujqësi përdoret si mjet që përshpejton procesin e pjekjes së frutave. Propileni dhe butilenet përdoren për të prodhuar polimere dhe alkoole të ndryshme. Por në prodhimin e gomës sintetike përdoret izobutileni. Prandaj, mund të konkludojmë se alkenet nuk mund të shpërndahen, pasi ato janë lëndët e para kimike më të rëndësishme.
Përdorimi industrial i etilenit
Në shkallë industriale, propyleni zakonisht përdoret për sintezën e polipropilenit dhe për prodhimin e izopropanolit, glicerinës, aldehideve butirik etj. Çdo vit rritet nevoja për propileni.

4.3.b. Shtimi i halogjeneve të hidrogjenit (hidrohalogjenimi)
Një reaksion tjetër i rëndësishëm i shtimit elektrofilik ndaj alkeneve është hidrohalogjenimi i njohur prej kohësh i alkeneve.
Më poshtë janë shembuj tipikë të shtimit të HCl, HBr dhe HI në alkene të ndryshme.
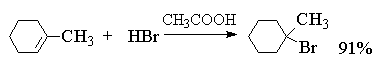

Efekti i zëvendësuesve të alkilit në lidhjen e dyfishtë në shkallën e shtimit përshkruhet nga sekuenca e mëposhtme:
R 2 C=CHR > RCH=CHR > RCH=CH 2
Kjo është në përputhje me mekanizmin në të cilin hapi përcaktues i shpejtësisë së reaksionit është formimi i një karbokation, pasi qëndrueshmëria e kationeve alkil zvogëlohet në serinë terciare > sekondare > parësore. Kështu, mekanizmi i shtimit duhet të përfshijë formimin e ndërmjetëm ose të një karbokacioni të lirë, që është i rrallë, ose të një ndërmjetësi me karakter karbokationik. Rasti i fundit është më tipik.
Nëse shtimi do të ndodhte përmes një "karbokacioni të lirë", atëherë reaksioni do të ishte plotësisht jostereoselektiv, pasi kationet alkil kanë një strukturë planare. Megjithatë, hidrohalogjenimi, si rregull, vazhdon në mënyrë stereoselektive, dhe në varësi të llojit të alkenit, selektiv anti- lidhje, selektive sin- ose të përziera sin-anti- aderimi.
Për alkenet, në të cilat lidhja e dyfishtë nuk është e konjuguar me një unazë aromatike, është karakteristikë anti- shtimi i një halidi hidrogjeni. Anti- shtimi i klorurit të hidrogjenit dhe bromit të hidrogjenit, klorurit të deuteriumit dhe bromit është vërejtur për cikloheksenin, ciklopentenin, 1,2-dimetilheksenin, 1,2-dimetilpentenin, cis- Dhe ekstazë-buten-2, heksen-3 dhe shumë alkene dhe cikloalkene të tjera të thjeshta.
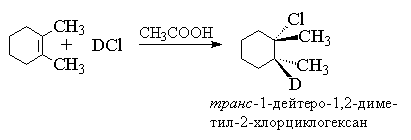
Në produktin shtesë, të njëjtët zëvendësues (grupe metil) ndodhen në anët e kundërta të rrafshit të mesëm të unazës së cikloheksanit, prandaj i përket ekstazë-rresht. Anti-shtimi vështirë se është i pajtueshëm me mekanizmin në të cilin supozohet formimi i një karbokacioni diskret. Për një karbokacion planar, sulmi nukleofilik i jonit halid është njësoj i mundshëm në të dy anët e planit, gjë që duhet të çojë në formimin e një përzierje produktesh sin- Dhe anti- lidhjet. Kinetika e hidrohalogjenizimit të alkenit gjithashtu tregon për një mekanizëm më kompleks të shtimit. Për alkenet jo të konjuguara, shpejtësia e reaksionit përshkruhet nga një ekuacion i rendit të tretë me një renditje të dytë për sa i përket halogjenit të hidrogjenit, d.m.th., ajo korrespondon me mekanizmin Ad E 3.
v = k [alken] 2
Reaksioni kundër shtimit dhe i rendit të dytë në lidhje me halogjenin e hidrogjenit është në përputhje të mirë me mekanizmin Ad E 3 në të cilin një alken reagon me dy molekula të halogjenit të hidrogjenit, njëra prej të cilave vepron si një elektrofil dhe tjetra si një agjent nukleofilik.

Një mekanizëm i tillë trimolekular sugjeron që fillimisht formohet një kompleks i një molekule alkeni dhe një molekule halide hidrogjeni, e ndjekur nga një sulm nga një molekulë e dytë HX në këtë kompleks me anti-anët pa formimin e një karbokacioni diskret. Duhet të theksohet posaçërisht se çdo mekanizëm trimolekular duhet të përbëhet nga dy faza të njëpasnjëshme, pasi përplasja e njëkohshme e tre molekulave është jashtëzakonisht e pamundur.

Kundër shtimit tregon një sulm nukleofilik preferencial të halogjenit të hidrogjenit nga ana e kundërt me atë nga e cila protonohet alkeni. Në vend të halogjenit të hidrogjenit, funksionin e agjentit nukleofilik në fazën përfundimtare mund ta kryejë edhe joni halid. Në të vërtetë, shpejtësia e reaksionit zakonisht rritet në përpjesëtim të drejtë me përqendrimin e jonit halid të futur në përzierjen e reaksionit në formën e halogjeneve tetraalkilamonium NR 4+ X - ose litium LiX. Në këtë rast, stereospecifike anti- aderimi.
Për alkanet, në të cilat lidhja e dyfishtë është e konjuguar me një unazë aromatike, është karakteristikë sin- lidhje ose e përzier sin-anti- shtimi i një halogjeni hidrogjeni, për shembull:
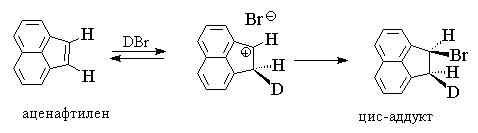
Syn-lidhja është procesi dominues për cis- Dhe ekstazë-izomerë të 1-fenilpropenit, 1-fenil-4-alkilciklohekseneve, acenaftilenit, indenit. Protonimi i alkeneve të tilla rezulton në formimin e karbokacioneve të tipit benzil, të cilët janë më të qëndrueshëm se kationet e pastër alkil që rezultojnë nga protonimi i alkeneve dhe cikloalkeneve konvencionale. Kinetika e reaksionit në këtë rast zakonisht përshkruhet nga një ekuacion më i thjeshtë i rendit të dytë v = k[alken], d.m.th., ajo korrespondon me mekanizmin bimolekular Ad E 2. Mekanizmi Ad E 2 përfshin formimin e një çifti jonesh duke përfshirë një karbokacion dhe një jon halide.

Nuk mund të pritet që mekanizmi i shtimit që përfshin çiftet e joneve të jetë shumë stereoselektiv. Nëse çifti jonik kthehet në produktin përfundimtar më shpejt se rrotullimi rreth një lidhjeje të thjeshtë karbon-karbon, rezultati përfundimtar do të jetë sin-shtimi, ku një proton dhe një jon halide janë ngjitur në të njëjtën anë të lidhjes dyfishe. Përndryshe, formimi i produkteve vërehet si sin- kështu dhe anti- Lidhjet HX. Një rast i tillë realizohet gjatë hidrohalogjenizimit çift-stirenet e zevendesuara Z-C 6 H 4 -CH=CH2 . Modeli i vërejtur këtu është ai sin- shtimi është tipik vetëm për ato olefina që, kur protonohen, japin një karbokacion relativisht të qëndrueshëm, d.m.th., në rastin e zëvendësuesve dhurues Z.
Reaksionet e hidrohalogjenizimit që zhvillohen sipas mekanizmit Ad E 2 karakterizohen nga konkurrenca midis proceseve të shtimit të konjuguar dhe rirregullimeve, pasi një karbokacion ose një çift jonik formohet si një ndërmjetës.

Si shembull i rirregullimeve me migrim 1,2 të një grupi alkil dhe një jon hidridi, ne paraqesim reaksionet e hidrohalogjenizimit të tert-butiletilenit dhe izopropiletilenit, përkatësisht.

Gjatë kryerjes së të njëjtit reaksion pa tretës në të ftohtë (-78 0 C), formohet një përzierje prej 33% produkteve shtese normale dhe 67% anormale (të rirregulluara).

4.3.c. Orientim. Rregulli i Markovnikov
Ndryshe nga elektrofilet simetrike (Hal 2), halogjenet e hidrogjenit janë reagentë elektrofilë josimetrikë. Shtimi i çdo elektrofili josimetrik (HBr, ICl, H 2 O, Hg (OAc) 2, etj.) në një alken josimetrik në parim mund të japë një përzierje të dy produkteve alternative, por në praktikë zakonisht formohet vetëm njëri prej tyre. Si shembull, merrni parasysh shtimin e bromurit të hidrogjenit në propilen.

Në vitin 1870, V.V. Markovnikov formuloi një rregull empirik sipas të cilit alkenet josimetrike shtojnë HX në mënyrë të tillë që formohet kryesisht një produkt në të cilin hidrogjeni shtohet në skajin më pak të zëvendësuar dhe X në skajin më të zëvendësuar të lidhjes dyfishe.
Zakonisht, rregulli i Markovnikov shpjegohet me ndryshimin në stabilitetin e dy karbokacioneve alternative. Për shembull, në shembullin e mësipërm, normalja n-Kationi i propilit është shumë më pak i qëndrueshëm se kationi izopropil, dhe për këtë arsye reaksioni vazhdon përgjatë rrugës së dytë.
Rregulli i Markovnikov fillimisht u përdor vetëm për shtimin e HX në substrate hidrokarbure, por në parim mund të shtrihet në reaksionet e alkeneve të tjera të zëvendësuara. Pra, shtimi i HCl në CF 3 CH=CH 2 jep " anti-Markovnikov "produkt CF 3 CH 2 CH 2 Cl. Kjo ishte e pritshme, pasi kationi CF 3 CH + CH 3 është më pak i qëndrueshëm se kationi CF 3 CH 2 CH 2 + për shkak të efektit të fortë (-I) të grupi CF 3 Kationi CF 3 CH 2 CH 2 + formohet kryesisht, por gjithashtu, megjithëse në një masë më të vogël, është i destabilizuar nga efekti induktiv i grupit CF 3, si rezultat i të cilit shtimi i HCl në trifluorometiletilen është shumë më i ngadalshëm se shtimi në etilen të pazëvendësuar.
Për një arsye të ngjashme, kationet e viniltrialkilamoniumit shtojnë HBr gjithashtu kundër rregullit Markovnikov:
Shtimi i HX në alkenet me zëvendësues të fortë (-I) dhe (-M), për shembull, tek akrilonitrili ose nitroetilen, duhet gjithashtu të shkojë kundër rregullit të Markovnikov. Megjithatë, në këtë rast lidhja e dyfishtë çaktivizohet aq fort në lidhje me reagentët elektrofilë, saqë këto reaksione zhvillohen vetëm në kushte shumë të rënda. Klorur vinil CH 2 =CHCl jep gjithmonë ekskluzivisht "addukte Markovnikov". Për shembull, kur ai reagon me HCl, formohet vetëm 1,1-dikloroetani (diklorid gjeminal) CH 3 CHCl 2. Klori, ngjashëm me grupin CF 3, ka një efekt të fortë (-I) dhe në pamje të parë duket se për këtë arsye shtesa duhet të ketë një orientim anti-Markovnikov, pasi kationi + CH 2 CH 2 Cl duhet të jetë më e qëndrueshme se kationi CH 3 CH + Cl. Megjithatë, ndryshe nga grupi CF 3, klori, përveç efektit (-I), ka edhe një efekt (+ M) që e kundërvepron atë (sepse ka çifte të vetme). Përvoja tregon se madhësia e efektit mesomerik është mjaft e mjaftueshme për të ulur energjinë e kationit 1-kloroetil nën nivelin e energjisë të kationit 2-kloroetil, në të cilin efekti +M nuk manifestohet.
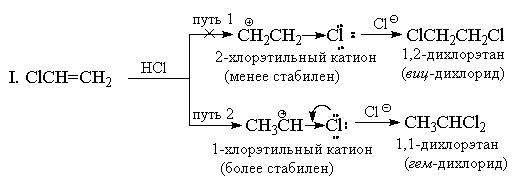
II. Nga pikëpamja e teorisë së rezonancës, struktura e kationit 1-kloroetil mund të përfaqësohet si më poshtë:
Megjithatë, shtimi i klorurit të vinilit ndodh më ngadalë sesa etilenit në të njëjtat kushte, d.m.th., sipas efektit total (-I> +M), klori mbetet një zëvendësues që tërheq elektronet në krahasim me hidrogjenin, dhe kationi 1-kloroetil është më pak e qëndrueshme se C 2 H 5 + . Halidet e tjera vinyl reagojnë në mënyrë të ngjashme me HX.
Eteret vinyl CH 2 =CHOR shtojnë HX (X=Hal) sipas rregullit të Markovnikov në një shkallë shumë më të lartë se të gjitha alkenet e zëvendësuara të listuara më sipër. Kjo është për shkak të efektit të rëndësishëm +M të grupit RO. Në kontrast me atomin e klorit, grupi RO për sa i përket efektit elektronik total (+M> -I) është një zëvendësues i fortë elektron-dhurues, duke stabilizuar në mënyrë efektive qendrën karbokationike fqinje. Struktura e karbokacionit në këtë rast mund të përfaqësohet gjithashtu si një grup i dy strukturave rezonancë
Sulmi i kationit të oksoniumit me një anion halidi çon në formimin e α-haloestereve të tipit CH 3 CH(Hal)OR.
4.3.d. Hidratimi i alkeneve
Hidratimi i alkeneve i katalizuar nga acidi çon në formimin e alkooleve. Drejtimi i hidratimit të alkeneve përcaktohet nga rregulli Markovnikov, kështu që supozohet se një karbokacion formohet si grimcë e ndërmjetme në këtë reaksion.
Tendenca e karbokacioneve dytësore të alkilit për t'u riorganizuar vetë pengon përdorimin e hidratimit të alkeneve për të marrë alkoole dytësore.

Kjo metodë në laborator ka gjetur një hapësirë të kufizuar vetëm për përgatitjen e alkooleve terciare. Reaksioni i hidratimit në këtë rast është kryesisht i kthyeshëm dhe alkoolet terciare formohen në rendimente të ulëta (40-45%).
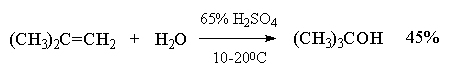

Hidratimi i alkeneve më të thjeshta - etilen dhe propileni - është një metodë e rëndësishme industriale për marrjen e alkooleve etilik dhe izopropil.

Në praktikën laboratorike, hidratimi i drejtpërdrejtë i alkeneve nuk ka gjetur aplikim të gjerë si për shkak të kushteve të vështira ashtu edhe për shkak të formimit të një sasie të konsiderueshme alkoolesh izomere. Aktualisht, reaksioni i lidhur me hidroksimerkurim-demerkurim përdoret zakonisht për prodhimin regioselektive të alkooleve nga alkenet.
4.3.d. Hidroksimerkurim-demerkurim
Sulmi elektrofilik në lidhjen e dyfishtë të një alkeni mund të kryhet nga jonet metalike, ndër të cilat kationi i merkurit (II) zë një pozicion të veçantë. Acetati i merkurit në kushte shumë të buta në 20 0 C u shtohet alkeneve në tetrahidrofuran ujor (THF) ose në acid acetik ujor për të formuar komponime organomerkuri. Shtimi i acetatit të merkurit në lidhjen e dyfishtë ndodh në mënyrë regjionale në përputhje të plotë me rregullin e Markovnikov, d.m.th., kationi i merkurit ngjitet në atomin e karbonit më pak të zëvendësuar.
Lidhja C-Hg në përbërjet organomerkuri mund të ndahet lehtësisht nga borohidridi i natriumit NaBH 4 për të formuar merkur dhe një lidhje të re C-H. Supozohet se hidridi i merkurit alkil përftohet si një ndërmjetës i paqëndrueshëm, i cili më tej dekompozohet me çlirimin e merkurit metalik nga një mekanizëm radikal.

Me pak fjalë, ky proces hidroksimerkurimi-demerkurimi me dy faza përfaqëson përfundimisht hidratim të alkenit regiospecifik sipas rregullit të Markovnikov në kushte jashtëzakonisht të buta, kur formimi i nënprodukteve reduktohet në minimumin maksimal të mundshëm. Kjo mund të ilustrohet qartë nga shembujt e mëposhtëm, në të cilët rendimenti total i produkteve të reaksionit është 90-98%. Të dhënat numerike të dhëna në këtë rast nuk tregojnë rendimentet e përbërjeve të formuara, por raportin e tyre në përzierje.




Siç mund të shihet nga shembujt e mësipërm, hidroksimerkurimi-demerkurimi i alkeneve në shumicën e rasteve siguron hidratim regiospecifik të alkeneve me formimin praktikisht vetëm të njërit prej dy alkooleve izomere. Duhet të theksohet se nuk ka nevojë të izolohet përbërësi organomerkur, dhe të dy proceset mund të kryhen drejtpërdrejt njëri pas tjetrit.
Hidroksimerkurimi i alkeneve josimetrike me sa duket fillon me sulmin e kationit AcOHg + dhe formimin e një kationi ciklik josimetrik të merkuriniumit (analog me jonin josimetrik të haloniumit) si një ndërmjetës, i cili më pas hapet si rezultat i një sulmi nukleofilik nga uji në atom karboni të zëvendësuar që mban një ngarkesë pozitive më të madhe.

Joni lidhës i merkurit mund të fiksohet në një mjedis jo-nukleofilik me aciditet të fortë edhe në 20 0 C duke shtuar një agjent elektrofilik më të fortë - trifluoroacetat merkuri në një përzierje të acidit fluorosulfonik dhe pentafluoridit të antimonit.

Kationi i merkurit mund të ndahet jo vetëm nga veprimi i ujit, por edhe nga tretës të tjerë elektron-dhurues: alkoolet, acidi acetik, acetonitrili, etj. Produkti përfundimtar i reaksionit në këtë rast do të jenë eteret, acetatet ose amide N-zëvendësuese të acid acetik, përkatësisht, për shembull:


Kur përdoren alkoole dytësore ose terciare të degëzuara në reaksionin alkoksimerkurim-demerkurim, Hg(OCOCF 3) 2 trifluoroacetate ose Hg (OSO 2 CF 3) 2 trifluoroacetate janë më efektive se acetati i merkurit.

Kështu, hidroksi- dhe alkoksimerkurimi-demerkurimi është një nga metodat më të mira përgatitore për sintezën e alkooleve dhe etereve me radikale alkile të degëzuara.
Shtimi i kripërave të merkurit tek alkenet është shembulli më i mrekullueshëm i një reaksioni të shtimit të konjuguar në një lidhje të dyfishtë, ku roli i agjentit të jashtëm nukleofilik luhet nga tretësi. Stereokimia e procesit të dyfishtë hidroksimerkurim-demerkurim varet nga rezultati stereokimik i çdo hapi individual. Hidroksimerkuracioni karakterizohet nga anti-shtimi, si për reaksionet e tjera që përfshijnë një kation ciklik. Megjithatë, demerkurimi radikal nuk karakterizohet nga stereoselektivitet i lartë. Prandaj, i gjithë procesi në tërësi është gjithashtu jostereospecifik.
4.3.f. Shtimi i alkeneve (dimerizimi kationik dhe polimerizimi i alkeneve)
Shembulli më interesant i këtij lloji të reaksionit është dimerizimi dhe polimerizimi i izobutilenit në prani të acidit sulfurik.

Emri teknik për këtë përzierje alkenesh është "diizobutileni". Ky reaksion zhvillohet sipas një mekanizmi kationik të ngjashëm me mekanizmin e shtimit të acideve minerale në lidhjen e dyfishtë të alkeneve. Në hapin e parë, një proton lidhet me një molekulë izobutileni për të formuar një molekulë relativisht të qëndrueshme. tert-butilkation. Formuar më tej tert-butilkacioni (acidi Lewis) reagon me një molekulë izobutileni (bazë Lewis) për të formuar një kation oktil terciar të ri të qëndrueshëm.

Në këto kushte, nën veprimin e bazave (joneve H 2 O, HSO 4 -), karbokacioni oktil humbet shpejt një proton dhe shndërrohet në një përzierje pentenesh izomere, pasi shkëputja e protonit ndodh nga dy pozicione të ndryshme:

Formimi preferencial i alkenit termodinamikisht më pak të qëndrueshëm - 2,4,4-trimetilpenten-1 (80% në përzierjen e reaksionit) shoqërohet me një akses më të madh hapësinor për sulmin bazë të atomeve të hidrogjenit të grupeve metil krahasuar me atomet e hidrogjenit. të grupit të metilenit. Në industri, "diizobutileni" hidrogjenizohet në Ni-Raney për të prodhuar "izooktan" (emri teknik për hidrokarburin 2,2,4-trimetilpentan), i përdorur si lëndë djegëse me oktan të lartë për motorët me djegie të brendshme.
Në përqendrime të larta të acidit sulfurik (më shumë se 80%), polimerizimi kationik i izobutilenit ndodh për të formuar një polimer të quajtur poliizobutileni (-CH 2 C(CH 3) 2 -) n . Ky polimer në formë gome përdoret për të marrë veshje anti-korrozioni dhe hidroizoluese, ngjitës, etj.
Përveç izobutilenit, sipas mekanizmit kationik polimerizohen 3-metilbuten-1, vinil eteret dhe disa derivate stiren të aftë për të formuar karbokacione relativisht të qëndrueshme. Fluori i hidrogjenit dhe acidet Lewis përdoren gjithashtu si katalizatorë për polimerizimin kationik: BF 3 , AlCl 3 , AlBr 3 dhe të tjera në prani të sasive shumë të vogla të ujit.
4.3.g. Shtimi i alkaneve (alkilimi i alkeneve)
Një metodë tjetër industriale për sintezën e "izooktanit" bazohet në ndërveprimin e izobutilenit me një tepricë të izobutanit në prani të acidit sulfurik të përqendruar ose në fluorid hidrogjeni anhidrik në 0 10 0 C.

Ky reagim vazhdon gjithashtu nëpërmjet mekanizmit kationik dhe, ajo që është me interes të veçantë, është një shembull i një procesi zinxhir kationik. Së pari, izobutileni dimerizohet në kushtet e reaksionit për të formuar një kation terciar oktil (CH 3) 3 CCH 2 C + (CH 3) 2 . Mekanizmi i formimit të tij përshkruhet në detaje në seksionin e mëparshëm. Më pas, ka një transferim të shpejtë të hidrogjenit (në formën e një joni hidridi) nga izobutani në kationin oktil me formimin e një molekule "izooktan" dhe një kation të ri tert-butil, i cili nga ana tjetër reagon shpejt me izobutilen për të formuar një kation i ri oktil etj.
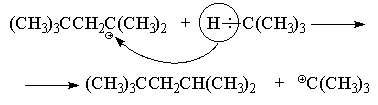
Krahas sintezës së "izooktanit", kjo metodë alkilimi përdoret në industrinë petrokimike për sintezën e hidrokarbureve të degëzuara me vlim të lartë nga alkenet e degëzuara dhe alkanet me zierje të ulët me një fraksion plasaritjeje termike.
ALKENET
Hidrokarburet, në molekulën e të cilave, përveç lidhjeve σ të thjeshta karbon-karbon dhe karbon-hidrogjen, ka lidhje π karbon-karbon quhen. e pakufizuar. Meqenëse formimi i një lidhjeje π është formalisht i barabartë me humbjen e dy atomeve të hidrogjenit nga një molekulë, hidrokarburet e pangopura përmbajnë 2p më pak atome hidrogjeni se kufiri, ku P - numri i lidhjeve π:
Një seri anëtarët e së cilës ndryshojnë nga njëri-tjetri me (2H) n quhet ana isologjike. Pra, në skemën e mësipërme, izologët janë heksane, heksene, heksadienet, heksinat, hekzatrienet etj.
Hidrokarburet që përmbajnë një lidhje π (d.m.th. lidhje e dyfishtë) quhen alkenet (olefinat) ose, sipas anëtarit të parë të serisë - etilenit, hidrokarburet e etilenit. Formula e përgjithshme për serinë e tyre homologe C p H 2n.
1. Nomenklatura
Në përputhje me rregullat e IUPAC, kur ndërtohen emrat e alkeneve, zinxhiri më i gjatë i karbonit që përmban një lidhje të dyfishtë merr emrin e alkanit përkatës, në të cilin mbarimi -en ndryshuar në -en. Ky zinxhir numërohet në atë mënyrë që atomet e karbonit të përfshirë në formimin e një lidhje dyfishe të marrin numrin më të vogël të mundshëm:

Radikalët emërtohen dhe numërohen si në rastin e alkaneve.
Për alkenet me strukturë relativisht të thjeshtë, lejohen emra më të thjeshtë. Pra, disa nga alkenet më të zakonshme quhen duke shtuar prapashtesën -en në emrin e një radikal hidrokarburi me të njëjtin skelet karboni:
Radikalet e hidrokarbureve të formuara nga alkenet marrin prapashtesën -enil. Numërimi në radikal fillon nga atomi i karbonit që ka një valencë të lirë. Sidoqoftë, për radikalët më të thjeshtë alkenil, në vend të emrave sistematikë, lejohet të përdoren ato të parëndësishme:

Atomet e hidrogjenit të lidhura drejtpërdrejt me atomet e karbonit të pangopur që formojnë një lidhje të dyfishtë shpesh quhen atomet e hidrogjenit të vinilit,
2. Izomerizmi
Përveç izomerizmit të skeletit të karbonit, në serinë e alkeneve ekziston edhe izomeria e pozicionit të lidhjes dyfishe. Në përgjithësi, izomerizmi i këtij lloji - Izomerizmi i pozicionit të zëvendësuesit (funksionet)- vërehet në të gjitha rastet kur në molekulë ka ndonjë grup funksional. Për alkanin C 4 H 10, dy izomerë strukturorë janë të mundshëm:
Për alkenin C 4 H 8 (buten), janë të mundshëm tre izomerë:

Buten-1 dhe butene-2 janë izomerë të pozicionit të funksionit (në këtë rast, roli i tij luhet nga një lidhje e dyfishtë).
Izomerët hapësinorë ndryshojnë në renditjen hapësinore të zëvendësuesve në lidhje me njëri-tjetrin dhe quhen cis izomere, nëse zëvendësuesit janë në të njëjtën anë të lidhjes dyfishe, dhe trans izomere, nëse në anët e kundërta:

3. Struktura e lidhjes së dyfishtë
Energjia e thyerjes së një molekule në lidhjen e dyfishtë C=C është 611 kJ/mol; meqenëse energjia e lidhjes σ C-C është 339 kJ / mol, energjia e thyerjes së lidhjes π është vetëm 611-339 = 272 kJ / mol. P-elektronet janë shumë më të lehta për t'u ndikuar sesa σ-elektronet, për shembull, nga tretësit polarizues ose nga ndonjë reagent sulmues. Kjo shpjegohet me ndryshimin në simetrinë e shpërndarjes së resë elektronike të σ- dhe π-elektroneve. Mbivendosja maksimale e p-orbitaleve dhe, rrjedhimisht, energjia minimale e lirë e molekulës realizohen vetëm me një strukturë planare të fragmentit të vinilit dhe me një distancë të shkurtuar C-C të barabartë me 0,134 nm, d.m.th. shumë më e vogël se distanca midis atomeve të karbonit të lidhur me një lidhje të vetme (0,154 nm). Me rrotullimin e "gjysmave" të molekulës në lidhje me njëra-tjetrën përgjatë boshtit të lidhjes dyfishe, shkalla e mbivendosjes së orbitaleve zvogëlohet, gjë që shoqërohet me shpenzimin e energjisë. Pasoja e kësaj është mungesa e rrotullimit të lirë përgjatë boshtit të lidhjes dyfishe dhe ekzistenca e izomereve gjeometrike me zëvendësimin përkatës në atomet e karbonit.
Alkenet- hidrokarbure të pangopura, të cilat përmbajnë një lidhje të dyfishtë. Shembuj të alkeneve:

Metodat për marrjen e alkeneve.
1. Plasaritja e alkaneve në 400-700°C. Reagimi zhvillohet sipas mekanizmit të radikalëve të lirë:

2. Dehidrogjenimi i alkaneve:
3. Reaksioni i eliminimit (ndarja): 2 atome ose 2 grupe atomesh shkëputen nga atomet fqinje të karbonit dhe krijohet një lidhje e dyfishtë. Këto reagime përfshijnë:
A) Dehidratimi i alkooleve (ngrohja mbi 150 ° C, me pjesëmarrjen e acidit sulfurik si një reagent që heq ujin):
B) Ndarja e halogjeneve të hidrogjenit kur ekspozohet ndaj një tretësire alkoolike të alkalit:

Atomi i hidrogjenit ndahet kryesisht nga atomi i karbonit që lidhet me një numër më të vogël atomesh hidrogjeni (atomi më pak i hidrogjenizuar) - sundimi i Zaitsev.
B) Dehalogjenimi:

Vetitë kimike të alkeneve.
Vetitë e alkeneve përcaktohen nga prania e një lidhjeje të shumëfishtë, prandaj, alkenet hyjnë në reaksione shtimi elektrofile, të cilat zhvillohen në disa faza (H-X - reagent):
Faza 1:

Faza e dytë:
 .
.
Joni i hidrogjenit në këtë lloj reaksioni i përket atomit të karbonit që ka një ngarkesë më negative. Shpërndarja e densitetit është:

Nëse ka një dhurues si një zëvendësues, i cili manifestohet si një efekt +I-, atëherë densiteti i elektronit zhvendoset drejt atomit më të hidrogjenizuar të karbonit, duke krijuar një ngarkesë pjesërisht negative mbi të. Reagimet vazhdojnë Rregulli i Markovnikov: kur bashkohen molekulat polare të tipit HX (HCl, HCN, HOH etj.) për alkenet josimetrike, hidrogjeni i shtohet preferencialisht atomit të karbonit më të hidrogjenizuar në lidhjen dyfishe.
A) Reaksionet e shtimit:
1) Hidrohalogjenimi:
Reagimi vazhdon sipas rregullit të Markovnikov. Por nëse peroksidi është i pranishëm në reagim, atëherë rregulli nuk merret parasysh:
2) Hidratimi. Reagimi zhvillohet sipas rregullit të Markovnikov në prani të acidit fosforik ose sulfurik:

3) Halogjenimi. Si rezultat, uji i bromit bëhet çngjyrosur - ky është një reagim cilësor ndaj një lidhjeje të shumëfishtë:
4) Hidrogjenizimi. Reaksioni zhvillohet në prani të katalizatorëve.


